
Accurate structure prediction of biomolecular interactions with AlphaFold 3
논문 링크 : https://www.nature.com/articles/s41586-024-07487-w
Nature에 엄청난 논문이 나왔었죠. Alphafold1이 나왔을 때 사람들이 엄청 놀랐는데, 벌써 세 번째 모델이 나왔습니다.
2024년 11월에 Publish된 해당 논문은 생성 모델이, 인공지능이 실제로 바이오 분야에 엄청난 도움이 된다는 것을 가장 잘 느끼게 해줍니다.
해당 논문 리뷰 전에 생성 모델의 기초가 되는 부분들을 리뷰해보면서 가봅시다.
Diffusion
우선 Diffusion 모델의 발전 과정과 개념에 대해서 리뷰를 하고 넘어가도록 하겠습니다.
diffusion 모델은 생성 모델의 한 종류로, 데이터에 점진적으로 잡음을 추가하는 forward process와 이를 역으로 제거하는 Reverse process 를 통해 새로운 데이터를 생성하는 기법입니다.

Forward Diffusion Process에서는 이미지에 고정된(fixed) 정규 분포(=Gaussian분포)로 생성된 Noise가 더해지고,
Reverse Diffusion Process에서는 이미지를 학습된(learned) 정규 분포로 생성된 Noise이미지로 뺍니다.
Diffusion Model이 풀려고 하는 문제는, Forward -> Reverse 단계를 거친 '결과 이미지'를 '입력 이미지'의 확률 분포와 유사하게 만드는 것 입니다. 이를 위해 Reverse단계에서, Noise 생성 확률 분포 Parameter인 평균과 표준편차를 업데이트하며 학습이 진행됩니다.
위 이미지는 2015년 Diffusion model에 해당하는 그림입니다. MLE(Maximum Log-Likelihood Estimation)를 사용한 결과이고, 아래의 그림은 유명한 연구인 DDPM에 해당됩니다. DDPM에서는 Reverse 단계에 U-net 모델을 사용했습니다.

사실 디퓨전 모델을 하나씩 자세학 리뷰하면 하나의 논문으로 거의 1시간을 채울 수 있을 만큼 수학적인 개념이 많이 요구됩니다. 그 중에서도 Markov chain과 Bayesian Rule은 수식을 이해할 때 가장 중요한 개념입니다.

생성 모델의 발전 과정
이제 Diffusion 모델의 발전 과정입니다.
Diffusion 모델의 전반적인 발전 흐름을 SCI급 저널과 최상위 컨퍼런스를 기준으로 선정한 핵심 논문들을 정리한 내용입니다.
2015년에는 확산 확률모델이 등장했습니다. 확산 기반의 생성 모델을 처음으로 제시했고, 이 논문에서는 비평형 통계역학이라는 개념을 활용하여 데이터의 구조를 서서회 파괴하는 forward process와 이를 거꾸로 진행시켜 데이터를 복원하는 reverse process 과정을 정의했습니다.
해당 논문의 가장 중요한 contribution은 이전의 GAN이나 VAE와 다른 제 3의 생성 모델 패러다임을 제시했다는 점입니다. 특히 이 방법은 수학적으로도 이점이 굉장히 많습니다. 예를 들어 명시적인 확률밀도 평가와 우도 계산이 가능하다는 장점이 있고, 얕은 신경망으로도 복잡한 데이터를 생성할 수 있다는 것을 보여줬습니다. 그리고 확산모델의 log likelihood를 계산해보면 다른 확률 모델 대비 경쟁력이 있다는 점도 확인했죠.
그래서 이 논문은 diffusion 모델 연구의 기초가 되었고, 후속 연구들은 이런 forward/reverse 프레임워크 위에서 다양한 개선과 변형을 시도하게 된 것이죠.
스코어 매칭(Score Matching) 방법을 활용한 새로운 생성 모델을 발표했습니다. 이 논문에서는 복잡한 확률분포를 직접 추정하는 대신,데이터 분포의 기울기(Score)를 추정하여 샘플을 생성하는 접근을 취합니다. 구체적으로, 데이터에 다양한 크기의 가우시안 노이즈를 섞은 변형들을 만들고, 각 노이즈 수준에서의 로그확률 기울기를 신경망으로 학습합니다.
그리고 Diffusion 모델 중 가장 중요한 연구 중 하나의 DDPM이 등장합니다.
DDPM(Denoising Diffusion Probabilistic Models)으로 불리는 확산 확률모델 입니다. 앞서 2015년 제안되었던 Diffusion 모델의 아이디어를 현대적으로 계승·발전시킨 이 논문은, 확산모델을 통해 당시 최고 성능의 이미지를 생성하며 큰 반향을 일으켰습니다. 특히 Score Matching 관점과 확산모델의 연결 고리를 이론적으로 제시하고, 효과적인 손실 최적화 기법을 알려준 것이 주요 공헌입니다.
DDPM 논문의 등장은 diffusion모델의 붐을 일으켰습니다. 이후 많은 연구가 DDPM의 구조와 손실을 개선하거나, 샘플링 속도를 높이거나, 조건부 생성에 적용하는 등으로 이어졌습니다. 대표적으로 OpenAI의 분석 논문 등이 나와 확산모델이 GAN을 대체할 강력한 후보로 부상하게 됩니다.
그리고 DDIM은 학습된 DDPM을 수정 없이 사용하면서도 상당히 적은 단계로 빠르게 샘플링할 수 있는 기법입니다. DDIM은 학습 절차를 바꾸지 않고도 확산모델의 느린 샘플링 문제를 상당 부분 개선했다는 큰 기여를 했습니다. 이후 확산모델을 응용할 때 DDIM이 자주 사용되어, 빠른 샘플 생성이나 이미지 편집(interpolation, morphing 등)에 활용되었습니다. 더 나아가 DDIM은 확산모델의 수학적 구조를 이해하는 데 기여하여, 확률적 확산을 ODE로 바라보는 통찰을 제공합니다.
정리하면, DDIM은 확산모델도 모수화에 따라 GAN처럼 결정론적 맵핑이 가능하다는 흥미로운 결과를 보여주었고, 실용적인 가속 이점까지 갖추어 확산 연구자들 사이에 널리 활용되고 있습니다.
| 연도 | 논문 | Contribution | 모델 특징 |
| 2015 | Deep Unsupervised Learning using Nonequilibrium Thermodynamics - ICML 2015 |
확산 확률 모델(diffusion probabilistic model)의 개념 제시. 순방향으로 데이터에 점진적으로 노이즈를 추가하고, 역방향으로 노이즈를 제거하며 데이터를 복원하는 마르코프 연쇄 생성 과정 제안. | Forward/Reverse 확산 프로세스 정의 |
| 2019 | Generative Modeling by Estimating Gradients of the Data Distribution | Score Matching을 활용한 새로운 생성모델 제안. 여러 수준의 가우시안 노이즈를 데이터에 첨가한 후 각 노이즈 수준별 스코어(확률밀도 기울기)를 신경망으로 추정 | Noise-Conditional Score Network (NCSN) 제안. |
| 2020 | Denoising Diffusion Probabilistic Models (DDPM) | - 확률적 확산 모델과 Score Matching의 연결 규명. - ELBO 기반 손실을 가중치 조정하여 단순화하고, 결과적으로 MSE 손실로 노이즈 예측 학습이 가능함을 제시. - CIFAR-10에서 Inception Score 9.46 / FID 3.17로 SOTA 달성, LSUN 256×256에서도 ProgressiveGAN에 필적하는 화질. |
|
| 2021 | Denoising Diffusion Implicit Models (DDIM) ICLR 2021 |
-확산 모델의 비마르코프(non-Markov) 샘플링 과정 제안.
-DDIM을 통해 샘플링 속도를 10~50배 가속하면서도 샘플 품질 저하가 미미함을 실증
|
네트워크 구조나 학습은 DDPM과 동일하지만, 생성시 T회가 아닌 훨씬 적은 단계로도 고품질 생성 |
뛰어난 이미지 품질에도 불구하고, 픽셀 공간에서 작동하는 Diffusion 모델은 계산량이 막대했습니다. 2022년 CVPR의 Rombach라는 사람이 이를 극복하기 위해 잠재 공간에서 확산을 수행하는 LDM (Latent Diffusion Model)을 선보였고, 이는 훗날 Stable Diffusion으로 알려졌습니다.
먼저 별도의 VAE(autoencoder)를 사용해 고해상도 이미지를 저차원 잠재 벡터 공간으로 압축합니다. 이 잠재공간에서는 이미지의 중요한 구조는 보존하면서도 차원이 크게 줄어들기 때문에, 확산모델의 연산부담이 감소. 이후 이 잠재 공간에서 DDPM과 유사한 확산모델을 학습하여, 잠재 벡터를 생성하도록 합니다. 최종적으로 생성된 잠재 벡터를 VAE 디코더로 변환해 이미지를 얻습니다. 이 접근으로, 픽셀 단위로 할 때 대비 수백 배의 속도 향상을 달성했습니다.
그리고 Cross-Attention을 도입했다는 것도 중요한 점입니다. 기존 diffusion U-Net은 클래스 레이블 등의 간단한 조건만 처리했는데, LDM은 Text, Bounding Box, Segmentation Map 등 임의의 조건을 처리하기 위해 U-Net 중간에 Cross-Attn 레이어를 넣었습니다. 예컨대 텍스트 조건의 경우, CLIP의 텍스트 임베딩 시퀀스를 Key, Value로 하고 U-Net의 feature를 Query로 하는 어텐션을 적용합니다. 이를 통해 거대한 텍스트 정보도 효과적으로 활용할 수 있게 되었고, 복잡한 조건부 생성 작업에 확산모델을 적용하는 길이 열렸습니다.
모델을 하나씩 다 자세하게 살펴보지는 않았지만 Diffusion 모델은 안정적인 확률적 생성 모델이고, 현재 이미지 생성 분야에서 가장 강력한 모델 중 하나로 자리 잡았습니다. 특히 저희가 연구하고 있는 메디컬 분야처럼 현실적 정확도와 다양성이 중요한 분야에서도 좋은 성과를 보이고 있고 앞으로도 지속적으로 연구와 응용이 활발할 것으로 기대됩니다.
| 연도 | 논문 | Contribution | 모델 특징 |
| 2022 | High-Resolution Image Synthesis with Latent Diffusion Models (Stable Diffusion) CVPR 2022 |
-고해상도 이미지 생성을 위해 잠재 공간(Latent Space)에서 확산을 수행하는 LDM 제안. 픽셀 공간에서 직접 확산하던 기존 방식의 연산량을 크게 절감하면서도 이미지 세부묘사 유지에 성공
-사전학습 VAE로 이미지→잠재벡터 변환 후, 그 잠재공간에 대해 확산모델 적용하여 복잡도 대 품질 절충의 최적점 도달
-또한 U-Net에 Cross-Attention 층을 도입하여 텍스트 등 임의 조건에 따른 이미지 생성을 자연스럽게 지원
-그 결과, 이미지 인페인팅에서 새로운 SOTA를 달성하고, 무조건 생성, 장면합성, 초해상화 등에서도 기존 대비 경쟁력 있는 성능을 보이며, 동일 성능시 계산 비용은 크게 감소됨.
|
VAE+Diffusion 구조 |
| 2022 | Photorealistic Text-to-Image Diffusion Models with Deep Language Understanding (Imagen) |
-대형 언어모델을 텍스트 인코더로 활용한 고해상도 텍스트-투-이미지 확산모델 제안
-DrawBench 벤치마크를 제안하여 타 모델(DALL-E2, GLIDE, LDM 등)과 비교한 결과, 인간 평가에서 Imagen의 텍스트-이미지 품질이 가장 우수함을 입증
|
- 거대 LM 결합 확산모델: 텍스트->임베딩에 파라미터 수십억 규모 |
Bioinformatics Data
제가 이 분야를 공부하기 시작하면서 가장 이해하기 힘들었던 부분이 데이터 타입에 관련된 부분이었습니다. 사실 다 같은 텍스트 데이터로 이뤄졌다고 생각했거든요. 현재 제가 연습하고 다루고 있는 데이터는 DNA와 RNA 같은 1차원 서열 정보를 주로 다루고 있습니다. 그런데 모두가 알고 있는 Alphafold나 RFdiffusion과 같은 생성 모델들은 모두 단백질 데이터를 다루고 있어요.
- DNA 데이터
- 보통 실험실에서는 특정 유전자(Gene) 또는 유전자들 일부를 PCR 증폭하거나, NGS로 짧게 잘라 시퀀싱한 후, 필요한 서열만 부분적으로 취급합니다.
- 즉, 전체 게놈 중 **“특정 관심 구간”**의 서열이 DNA 데이터가 될 수 있습니다.
- 이중나선 형태가 일반적이지만, 분석할 때는 일단 1가닥(5’→3’ 방향 서열)으로 표현하는 경우가 많습니다.
- RNA 데이터
- mRNA, miRNA, lncRNA 등 다양한 종류가 존재하며, 보통 단일가닥(Single-stranded) 구조.
- 2차 구조(부분적 스템-루프, 염기쌍 형성)와 3차 구조가 존재할 수 있으나, DNA에 비해 역동적이고 변형이 많습니다.
- RNA-Seq(NGS)를 통해 세포 내에서 발현된 전사체(Transcriptome)를 “짧은 리드” 형태로 얻은 뒤, 이를 어셈블리하거나 유전자의 mRNA 발현량을 측정하는 용도로 사용합니다.
- 스케일의 차이
- Genome은 전체 규모가 매우 큰 반면, 연구자가 실제로 분석·조작하려는 DNA/RNA는 특정 유전자(수 kb) 또는 특정 영역 중심으로 접근하는 경우가 많습니다.
- RNA의 경우, 단백질 코딩 길이가 짧으면 수백
수천 bp(mRNA), 비코딩 RNA는 수십수백 nt 정도로 짧기도 합니다.
- 구조적 특징
- DNA: 디옥시리보스(2’-H) 당, 염기 A/T/G/C, 2중가닥이 기본.
- RNA: 리보스(2’-OH) 당, 염기 A/U/G/C, 주로 단일가닥(부분적 이중가닥 형성 가능), 더 다양하고 역동적인 입체구조.
| 구분 | DNA | RNA | 단백질 (Protein) |
| 생물학적 역할 | 유전 정보 저장, 전달 | 유전 정보 전달, 유전자 발현 조절 및 단백질 합성 중간 매개 | 세포 내 생명 현상 직접 수행 (효소작용, 신호전달, 구조적 역할 등) |
| 기본 구성 단위 | A, T, G, C | A, U, G, C | 20종의 아미노산 (Amino Acid) |
| 데이터 구조적 특징 | 1차원 서열 정보 | 1차원 서열 정보 | 1차원 서열 및 3차원 입체 구조 |
| 생물학적 데이터 예시 | Genome 시퀀스 데이터 | - RNA-seq (발현량 데이터) - 전사체(transcriptome) |
- 아미노산 서열 - PDB 단백질 구조 데이터 |
| 데이터 분석 목적 |
-변이 분석
-유전체 연관 연구(GWAS)
-SNP 분석
-유전자 발굴
|
- 유전자 발현 분석 - Alternative splicing - RNA 변형 분석 - 질병 연관성 분석 | - 단백질 구조 예측 -기능 예측
-약물 결합 예측
-구조 기반 약물 설계
|
왜 Genome·단백질 수준에서는 생성 모델 연구가 활발하고, DNA/RNA 수준은 상대적으로 덜 활발한가?
단백질 구조·설계 분야의 배경
- 풍부한 구조 데이터(PDB 등)
- Protein Data Bank(PDB)에 축적된 단백질 3D 구조가 19만 건 이상 존재하고, AlphaFold2 등으로 예측된 구조도 대규모 공개됨.
- 이렇게 쌓인 방대한 3차원 구조 데이터가 구조 기반 생성 모델(RF Diffusion 등)을 훈련하는 데 큰 자원이 됩니다.
- 의약·산업적 수요
- 의약품 타겟 또는 효소 설계 등, 단백질 구조 설계의 산업적 가치가 매우 높습니다.
- 따라서 단백질 생성 모델(특히 구조·기능 설계)은 투자가 많고, 빠른 발전을 보이게 되었습니다.
- 상대적으로 명확한 품질 평가 지표
- 단백질의 “접힘(folding) 안정성”, “결합 에너지”, “효소 반응성” 등 물리·화학적으로 정의된 스코어(Rosetta 등)가 존재.
- 모델이 생성한 구조를 평가하고 피드백하기 용이하여, Diffusion 모델을 포함한 생성적 접근이 빠르게 발전했습니다.
단백질(Protein)에 집중되는 이유
- 방대한 구조 데이터(PDB 등) 확보
- 실험적으로 결정된 단백질 3D 구조 + AlphaFold2 예측 구조가 대규모로 공개되어 있습니다.
- 3차원 구조가 명확하게 정리·표준화된 형태로 쌓여 있어, RF Diffusion 등 생성 모델 학습에 큰 자원이 됩니다.
- 의약·산업적 가치는 매우 높음
- 단백질은 효소, 항체, 의약품 타깃으로 산업적 수요가 막대합니다.
- 투자·연구가 활발하며, 새로운 단백질(기능/안정성 향상 등)을 설계·생성하는 연구 필요가 큽니다.
- 명확한 물리·화학적 품질 평가 지표
- 접힘(folding) 안정성, 결합 에너지, 효소 활성도 등 측정 가능한 스코어가 존재합니다.
- 모델이 생성한 구조를 Rosetta 등으로 검증해 피드백 루프를 빠르게 돌릴 수 있어, 연구 개발이 용이합니다.
- 단백질 3차원 구조에 잘 맞는 Diffusion 기법
- 단백질은 고정된 형태로 폴딩되므로, 3D 좌표(백본, 원자) 기반의 확산 모델을 적용하면 곧바로 의미 있는 결과를 얻을 수 있습니다.
- 이 점이 RF Diffusion의 빠른 발전으로 이어졌습니다.
- 단백질은 풍부한 3D 구조 데이터, 명확한 물리·화학적 평가 지표, 높은 산업적 가치 때문에 Diffusion 모델을 비롯한 생성 모델이 빠르게 성장했습니다.
- 반면 DNA/RNA는 1차원 서열 + 복합적 기능, 구조 데이터 부족, 검증 난이도, 시장 수요 등의 제약으로 인해, 아직은 단백질만큼 Diffusion 모델 연구가 폭발적으로 진행되지 못한 상황입니다.
- 그러나 mRNA 치료제, 합성생물학 등 분야가 발전함에 따라, DNA/RNA 확산 모델도 앞으로 점차 주목받고 연구가 늘어날 가능성이 높습니다.
우선 Bioinformatics 분야에서는 어떤 데이터를 다루는지 알아보겠습니다.
표를 보면 DNA RNA는 주로 1차원 시퀀스 기반 데이터를 다루며, 유전자 발현 및 변이를 통해 생물학적 현상을 분석합니다. 단백질은 1차원 시퀀스 뿐만 아니라 3차원 입체 구조 정보가 매우 중요하고 구조와 기능이 강력한 상관관계를 갖습니다.
Alphafold3
알파폴드 1,2 는 간략하게 말씀드리면 1차원 서열로 구성된 단백질이 입력으로 들어왔을 때, 이것들이 어떻게 접히면서 어떤 구조가 되는지 예측하는 수준에 머물렀다면 이번에 알파폴드3는 구조를 예측하는 것 뿐만 아니라 단백질이 어떤 DNA와 결합할 수 있는지까지 예측하는 그런 조금 더 업그레이드 된 모델이라고 볼 수 있습니다.
알파폴드 1,2의 경우 단백질이 어떤 분자로 구성되어 있는지에 대한 설계도를 주면 단백질이 이러한 형상으로 생겼을 것이다 라고 예측 모델로 예측을 해주는 것입니다. 그런데 단백질 구조만 예측하는 것이 중요한게 아니라 어떤 DNA와 어떻게 상호작용 하는지 혹은 단백질이 리간드와 결합되었을 때 어떻게 변하는지에 대해서도 신약 개발에 있어서 매우 중요합니다. 알파폴드 3가 이제 그런 역할도 할 수 있는 모델이라고 보시면 될 것 같습니다.
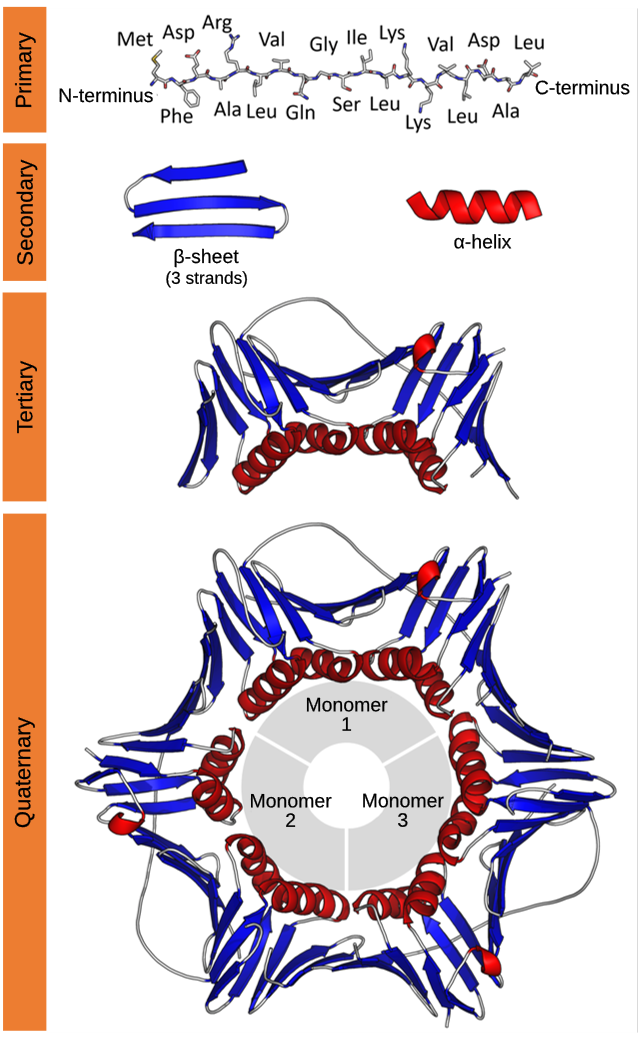
우선 단백질의 구조에 대해서 알아야 하는데요
사진의 가장 위에 있는 것을 보시면 아미노산이 1차원적으로 연결되어 있습니다. (primary)
저기에서 아미노산을 제가 원하는 종류로 바꿀 수 있습니다. 그리고 실제 상온이나 물에 넣게 되면 저 구조가 접히기 시작해요. 아래에 보면 2차구조 secondary처럼 저렇게 바뀌게 됩니다.
저런 2차 구조들이 여러가지 그룹으로 묶이게 되면서 더 복잡한 3차 구조(terti)가 만들어져요. 이런 3차 구조들이 모여서 마지막에 프로틴 구조인 마지막 모습이 됩니다.
그래서 첫 번째부터 마지막 단계까지 예측하는 모델이 알파폴드 1이랑 2 였습니다.
알파폴드3의 경우에는 저러한 단백질과 인풋 값으로 다른 DNA 혹은 리간드를 넣을 때 어떻게 결합하는지를 밝혀내는 일을 합니다.
Alphafold3 Architecture
단백질 구조나 생명 공학에서의 시사점 등 해당 논문에 대해서는 많은 의견이 있습니다. 하지만 이번 발표에서는 머신러닝 관점에서 알파폴드의 모델에 대해서 리뷰하려고 합니다.
우선, 모델의 목표가 이전 AlphaFold 모델과는 조금 다르다는 점에 주목할 필요가 있습니다. AlphaFold2(AF2)는 단백질 단일 서열에 대한 3차원 구조를 예측했고, AlphaFold-Multimer는 여러 단백질이 복합체를 이루는 구조를 예측했습니다. 반면 AlphaFold3(AF3)는 단백질뿐 아니라, 다른 단백질·핵산(DNA/RNA)·소분자 등이 함께 존재하는 복합체 구조도 서열 정보만으로 예측합니다.
이전 AF 모델들은 표준 아미노산(20가지)만 다루면 됐지만, 이제 AF3는 훨씬 다양한 입력 타입(예: 핵산 염기, 비표준 잔기, 소분자 등)을 표현해야 하므로, 더 복잡한 피처화/토큰화 방식을 갖습니다. 해당 토큰화 방식을 별도 섹션에서 다루겠지만, 여기서는 일단 “토큰(token)은 단백질의 경우 단일 아미노산, 핵산의 경우 단일 뉴클레오타이드, 혹은 (표준 아미노산/뉴클레오타이드에 속하지 않는) 개별 원자를 의미한다”고 생각하시면 됩니다.
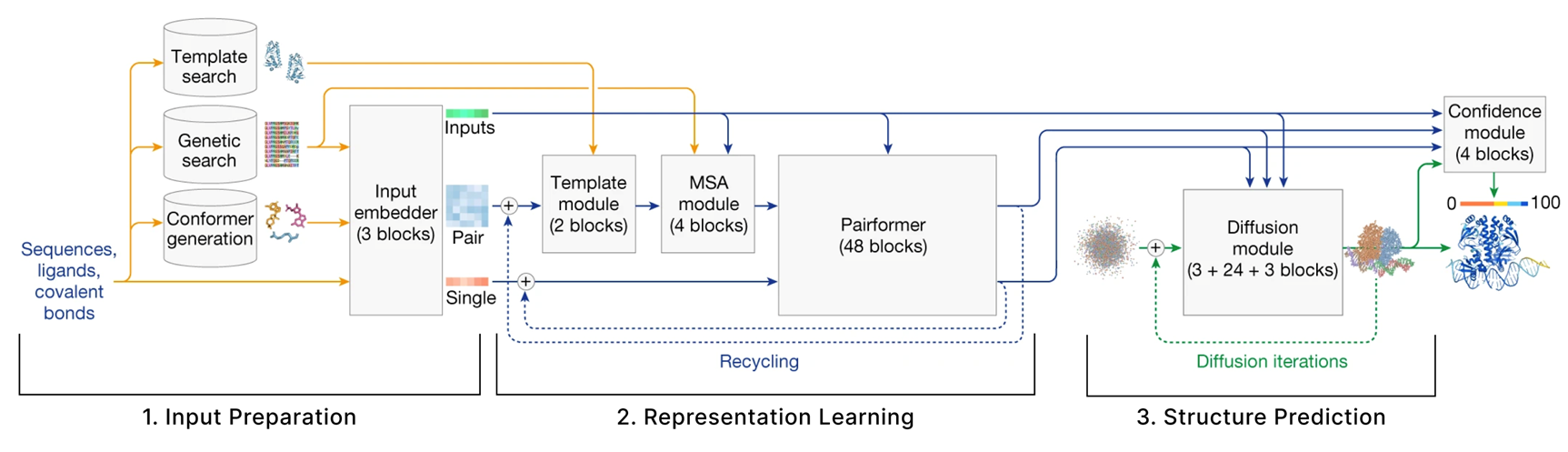
모델은 크게 세 가지 섹션으로 나눌 수 있습니다.
- 입력 준비 (Input Preparation)
- 사용자가 예측하고자 하는 분자들의 서열을 제공하면, 이를 숫자 텐서 형태로 임베딩해야 합니다.
- 또한, 모델은 사용자가 제공한 분자들과 유사한 구조를 지닐 것으로 추정되는 다른 분자들의 정보를 검색해서, 이들도 자체적으로 텐서로 임베딩합니다.
- 표현 학습 (Representation Learning)
- 앞 단계에서 생성된 Single 텐서와 Pair 텐서를 입력으로 받아, 다양한 형태의 어텐션 기법을 적용하여 이들 표현을 업데이트합니다.
- 구조 예측 (Structure Prediction)
- 업데이트된 Single/Pair 표현과, 1단계에서 준비된 원본 입력을 바탕으로 **조건부 확산(conditional diffusion)**을 사용해 최종 구조를 예측합니다.
Single & Pair
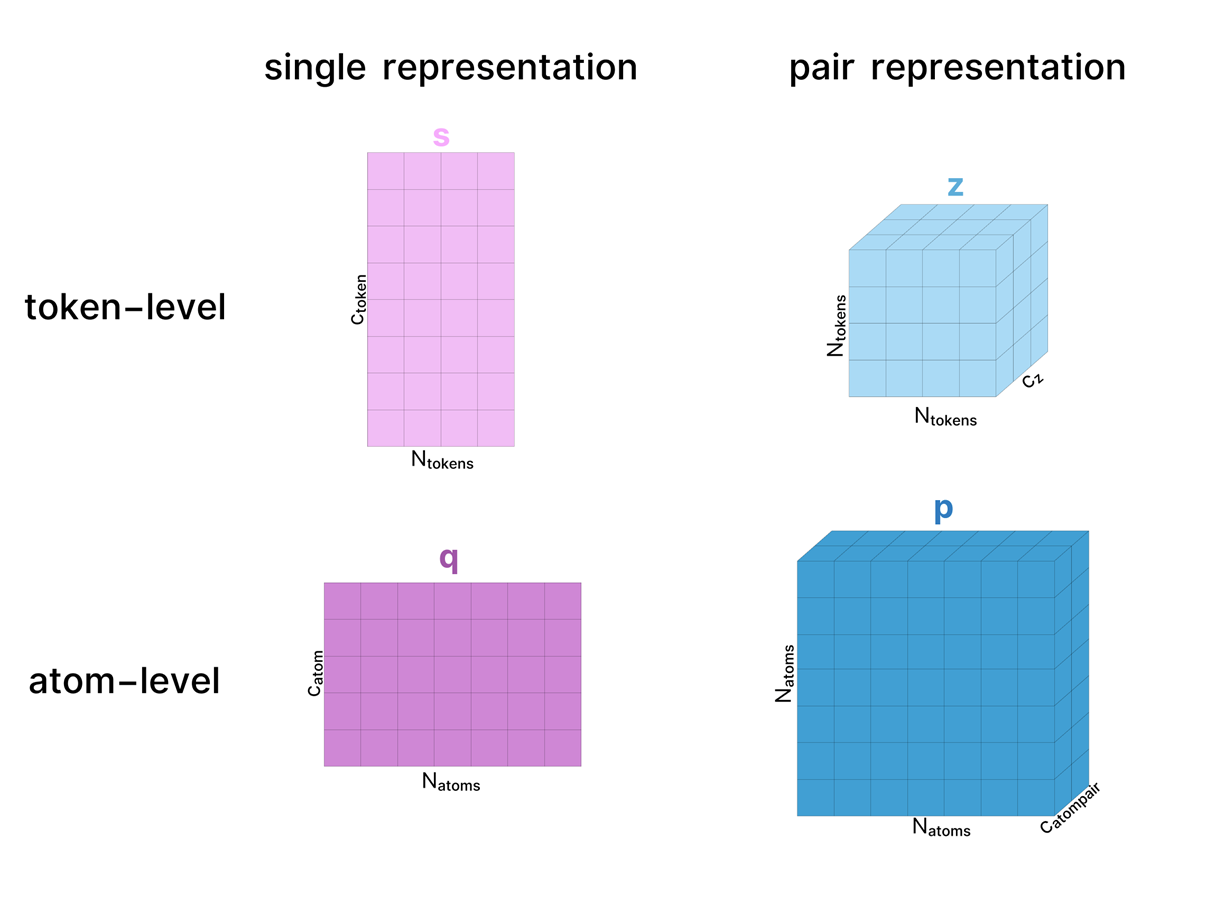
AF 모델 전반에서 단백질 복합체는 크게 두 가지 형태로 표현됩니다.
- Single 표현 : 복합체 내 모든 ‘토큰’을 나타냅니다. (ex.아미노산, 원자)
- pair 표현 : 복합체 내 아미노산(또는 원자) 쌍 사이의 관계(ex. 거리, 잠재적 상호작용 등)를 표현합니다
이 각각은 원자(atom) 단위나 토큰(token) 단위 중 하나로 다룰 수 있으며 AF3 논문에서 정의된 이름과 색상을 사용해서 저렇게 시각적으로 구분해서 사용할 예정입니다.
가중치(Weights)는 생략하고, 실제로는 활성값(Activation)의 형태 변화만 시각화합니다.
각 활성 텐서에는 논문에서 사용한 차원 이름을 그대로 라벨링하며, 다이어그램의 크기도 (정확하진 않더라도) 해당 차원의 증가/감소를 대략적으로 반영하도록 그렸습니다. 가능하면, (이 다이어그램뿐 아니라 모든 다이어그램에서) 텐서 위에 표시된 이름이 AF3 보충자료에서 쓰인 텐서 이름과 동일하도록 유지했습니다.
보통 텐서는 모델을 거치면서 같은 이름을 유지하지만, 어떤 경우에는 처리 단계별로 다른 버전을 구분하기 위해 이름을 달리 쓸 때도 있습니다. 예를 들어, 원자 단위 single 표현에서는 초기 표현을 c라 하고, Atom Transformer를 거쳐 업데이트된 표현을 q라 부르는 식입니다.
설명을 단순화하기 위해, 도식에 등장하는 대부분의 LayerNorm은 생략했으나, 실제 모델에서는 거의 모든 단계에서 사용됩니다.
사용자가 AF3에 입력하는 실제 데이터는 기본적으로 하나의 단백질 서열이며, 필요에 따라 다른 분자들(핵산, 소분자 등)을 추가로 제공할 수 있습니다. 본 섹션의 목표는 이들 서열을 아래 그림과 같이, 모델 메인 트렁크로 들어가기 위한 6가지 텐서로 변환하는 것입니다. 이 텐서들은 주석을 달아놓은 것과 같습니다.
이 섹션의 최종 결과로 원자(atom) 단위 표현(q, p)뿐 아니라, 해당 원자 정보를 집계(aggregation)하여 만든 토큰(token) 단위 표현(s, z)까지 모두 생성됩니다. 즉, 이 과정이 끝나면 모델은 Single(단일) 표현과 Pair(쌍) 표현을 각각 원자 수준과 토큰 수준 두 가지 형태로 모두 얻게 됩니다.
지금 보시는 이 섹션은 총 5가지로 나뉘게 됩니다.
•s: 토큰 단위 단일(single) 표현 (token-level single representation)
•z: 토큰 단위 쌍(pair) 표현 (token-level pair representation)
•q: 원자 단위 단일(single) 표현 (atom-level single representation)
•p: 원자 단위 쌍(pair) 표현 (atom-level pair representation)
•m: MSA(다중 서열 정렬) 표현 (MSA representation)
•t: 템플릿(template) 표현
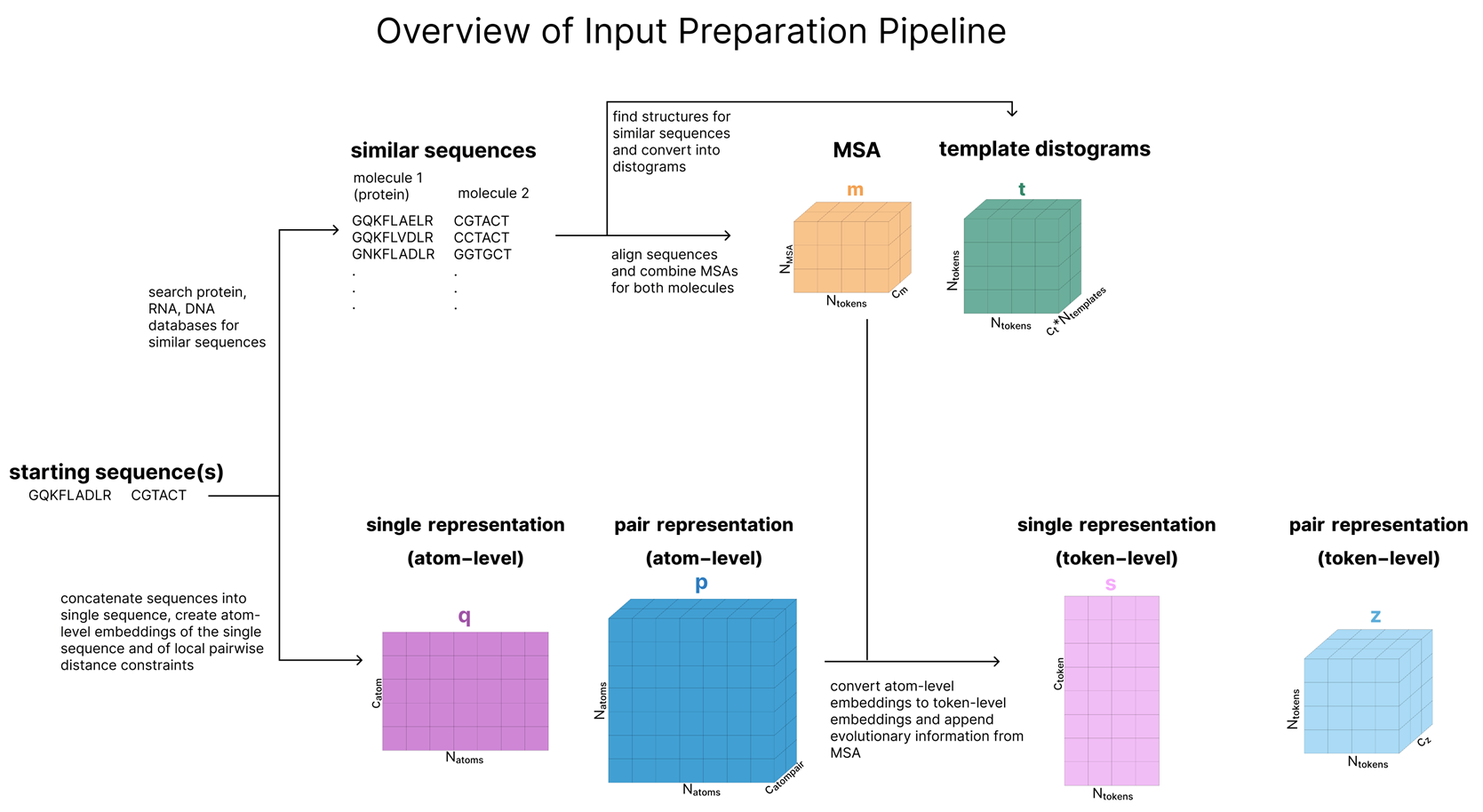
Tokenizer
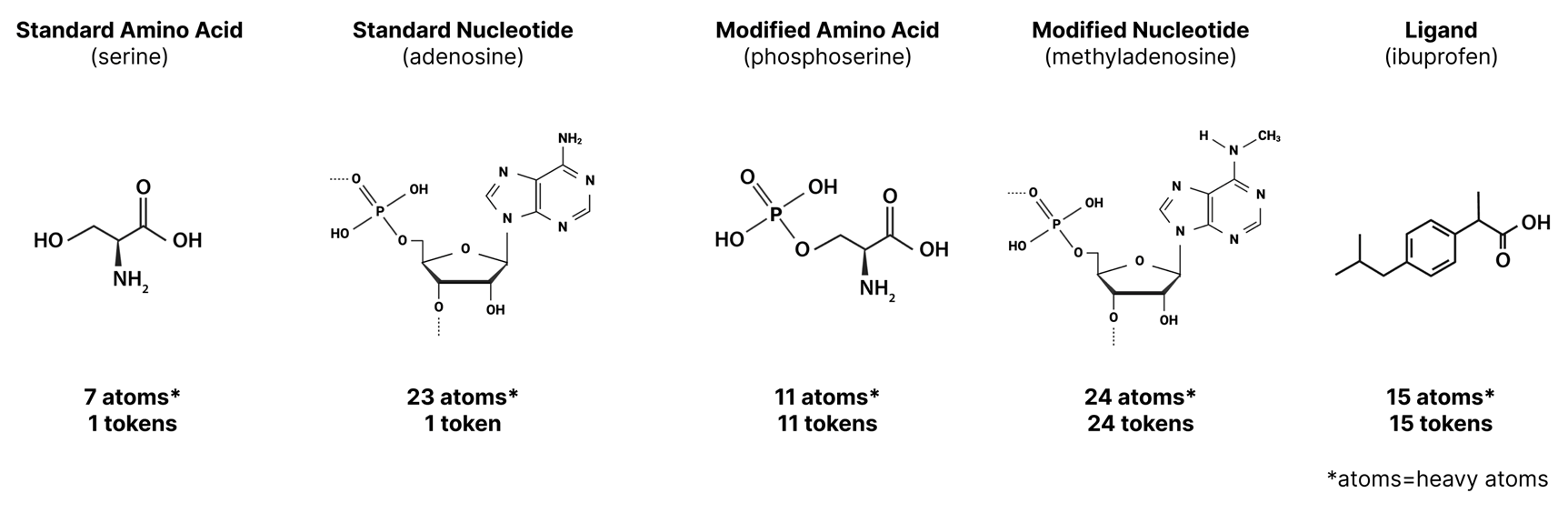
첫 번째는 Tokenizer 입니다.
AF2에서는 고정된 아미노산 세트만 모델이 다뤘고, 각 아미노산마다 토큰 하나를 할당했습니다. 이 방식은 AF3에서 그대로 유지되지만, AF3가 처리할 수 있는 분자 유형이 늘어났기 때문에 추가 토큰들이 도입되었습니다.
- 표준 아미노산: 1 토큰 (AF2 방식과 동일)
- 표준 뉴클레오타이드(DNA/RNA 염기): 1 토큰
- 비표준 아미노산/뉴클레오타이드(메틸화된 뉴클레오타이드, 번역 후 변형이 일어난 잔기 등): 원자별로 각각 1 토큰
- 그 외 분자(리간드, 이온 등): 원자별로 각각 1 토큰
이로 인해, 어떤 토큰은 여러 개의 원자(예: 표준 아미노산)와 연결되지만, 다른 토큰은 단 하나의 원자(예: 리간드 내 특정 원자)에만 대응될 수 있습니다. 예를 들어, 표준 아미노산이 35개 들어 있는 단백질(아마도 600개 이상의 원자가 존재)이라면, 이 단백질은 토큰 35개로 표현됩니다. 반면 35개의 원자를 가진 어떤 리간드는, 각각의 원자에 1토큰씩 할당되어 역시 35개 토큰이 됩니다.
Retrieval (Create MSA and Templates)
AF3에서 매우 초기 단계에 수행되는 핵심 작업 중 하나는, 마치 언어 모델의 RAG(Retrieval Augmented Generation)와 유사한 방식으로, 우리의 관심 대상인 단백질 또는 RNA 서열과 유사한 서열을 찾아서 (MSA라 부르는 다중 서열 정렬에 모으고), 거기에 대응하는 구조 정보(“템플릿”)가 있으면 이를 추가 입력으로 활용하는 것입니다.
그렇게 찾은 서열과 구조가 모델에서 각각 m(MSA), t(템플릿)라는 입력으로 주어집니다.
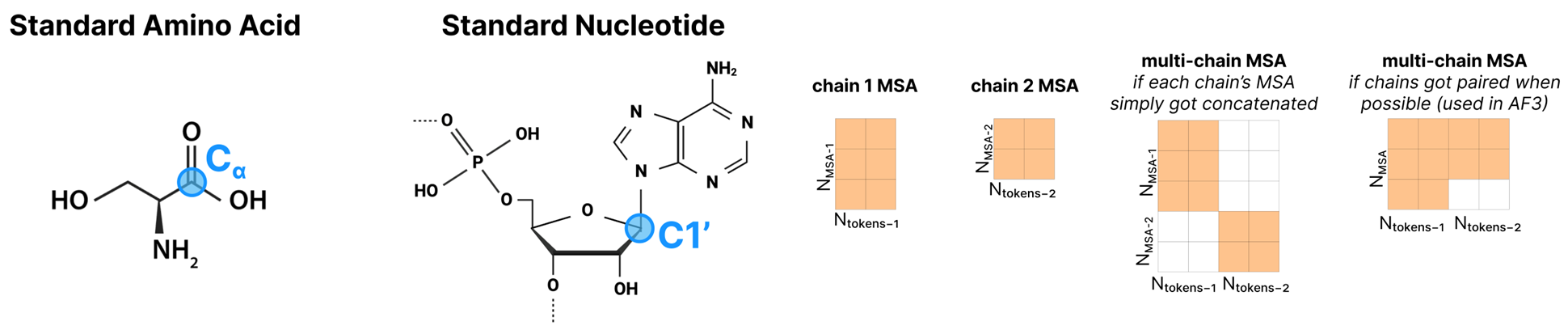
MSA(다중 서열 정렬)의 이점
서로 다른 종(種)에 존재하는 상동(homologous) 단백질들은 서열·구조 면에서 상당히 유사한 경우가 많습니다. 단백질 하나의 특정 위치(열, column)가 진화 과정에서 어떻게 바뀌었는지 보면, 그 위치에 어떤 아미노산이 중요한지, 혹은 서로 상호작용하는 아미노산들이 어떻게 공변(covary)하는지를 알 수 있습니다. 이미 AlphaFold2 시절부터, MSA가 단백질의 단일 서열에 비해 훨씬 풍부한 ‘보존·공변 정보’를 제공한다는 것이 밝혀졌습니다.
구조 템플릿(template)의 활용
유사한 서열을 지닌 단백질에 대해 이미 결정된 실험 구조가 있다면, 이는 해당 단백질(우리가 예측하고자 하는 단백질)의 구조를 추론하는 중요한 단서가 됩니다. 완전한 전체 구조를 찾는 대신, 단백질 체인 하나씩 분리하여 유사도를 검사하고, 이 중 상위 품질 구조 4개 정도를 추려 “템플릿”으로 삼습니다. 이는 전통적인 호모로지 모델링(homology modeling)과 유사한 방식입니다.
템플릿은 어떻게 표현되는가?
- 중심 원자(center atom) 기준 거리 행렬
- 템플릿 구조가 확보되면, 해당 체인의 각 토큰(아미노산 or 뉴클레오타이드 등)에 대해 “중심 원자”(Cα 혹은 C1’)를 골라,
- 각 토큰 쌍 사이 유클리드 거리를 계산합니다.
- 이렇게 얻은 N_token x N_token 거리 행렬을 연속값 대신 히스토그램(bin) 형태로 구분해 **“distogram”**으로 만든 뒤, 모델 입력에 넣습니다.
- 메타데이터 포함
- 각 토큰이 어느 체인에 속하는지, 실제 결정 구조에서 이 토큰(잔기)이 해석되었는지, 아미노산 내부 지역 거리(백본 길이 등) 같은 정보도 추가.
- 단, 템플릿은 체인 간 상호작용 정보를 포함하지 않으며(체인 A-B 거리 무시), 단일 체인 내 거리에 집중합니다
- 요약하자면, AF3는 “(1) 서열 기반 MSA 구성 → (2) 유사 구조 템플릿 선택” 과정을 통해, 부가 정보를 풍부히 확보합니다.
- 이것이 바로 m(MSA), t(템플릿)으로 모델에 입력되며, 단백질+RNA 복합체든 멀티머든 보다 정확한 구조 예측을 지원하게 됩니다.
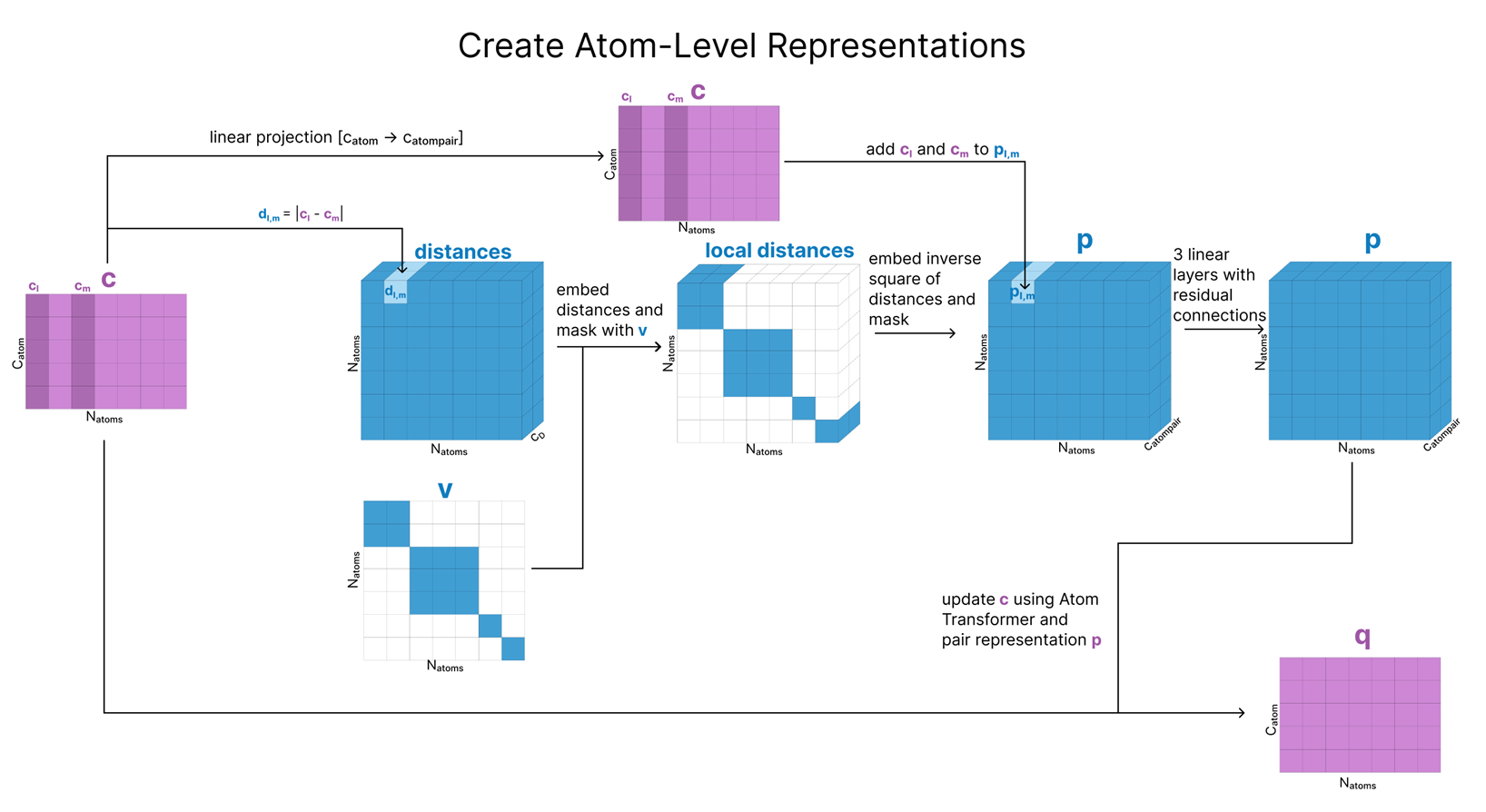
Create Atom-Level Representations
q(원자 단위 single 표현)을 만드는 과정은 다음과 같은 단계를 거칩니다:
- 참조 컨포머(reference conformer) 생성
- 단백질, 핵산, 리간드 각 구성성분에 대해, 미리 알려진 부분 구조(“로컬 구조”에 대한 사전 지식)를 활용해 3D 컨포머를 얻습니다.
- 아미노산의 경우: 이미 에너지가 낮은(안정된) 컨포머가 “표준” 형태로 정해져 있어, 룩업(look-up) 방식으로 불러올 수 있습니다.
- *소분자(리간드 등)**의 경우: RDKit의 ETKDGv3 알고리즘을 이용해 실제 단일 결합 회전 등에 기초한 3D 컨포머를 생성합니다.
- 원자 단위 특징정보 취합
- 이렇게 얻은 컨포머에서, 각 원자의 상대 위치(좌표)와 전하, 원자번호, 기타 식별 정보 등을 합쳐서 행렬 c에 저장합니다.
- 요약하면, c에는 “원자별 공간좌표 + 물리화학 속성”이 들어 있다고 보면 됩니다.
- 원자 단위 pair 표현 p 초기화
- 원자들 간 상대 거리 정보를 p에 저장하는데, 컨포머를 통해 알 수 있는 거리만 초기화하고,
- 아직 알 수 없는 거리(토큰 간 상호작용 등)는 마스크( v )로 처리하여 배제합니다(초기 단계에서는 “몰라”로 두는 셈).
- 거리의 역제곱(inverse square)에 대한 임베딩을 적용하고, cl, cm(추가 특징) 등을 투영(projection)한 뒤, 몇 개의 Linear layer + Residual connection을 거쳐 p를 갱신합니다.
- 원자 단위 single 표현 q 복사 생성
- 초기 행렬 c를 복사해 이름을 q라고 붙입니다.
- 이후 q가 곧바로 업데이트될 주된 원자 단위(single) 표현이며,
- c는 나중에 참조용으로 다시 쓰이므로, 삭제되지 않고 저장해 둡니다.
정리하면, 각 분자(아미노산·핵산·소분자)의 참조 컨포머를 활용해 원자별 좌표·속성을 모으고, 이를 기반으로 원자 단위 pair 표현(p)를 초기화하며, 그 과정에서 생성된 정보를 q로 복사해서 이후 단계에서 계속 업데이트한다는 흐름입니다.
컨포머(conformer)란, 분자의 원자 간 연결 상태(결합) 자체는 동일하지만, 단일 결합(single bond)을 중심으로 한 회전 각도가 달라져서 생기는 서로 다른 3차원 구조를 말합니다.
- 예: 에탄(탄소-탄소 단일 결합)처럼 단일 결합을 가진 분자는, 그 결합을 축으로 원자가 회전해 여러 가지 3D 배치가 가능하고, 이 각각이 곧 ‘컨포머’입니다.
- 보통 분자는 결합 길이·각도 등 큰 변화 없이, 단일 결합의 회전만으로도 3D 형태가 달라질 수 있기 때문에, 에너지가 낮은(안정된) 특정 배치를 찾아 “표준 컨포머”로 사용하거나, 시뮬레이션에서 다양한 컨포머를 생성해 어떤 구조가 가장 안정적인지 탐색하기도 합니다.
Update Atom-Level Representations (Atom Transformer)
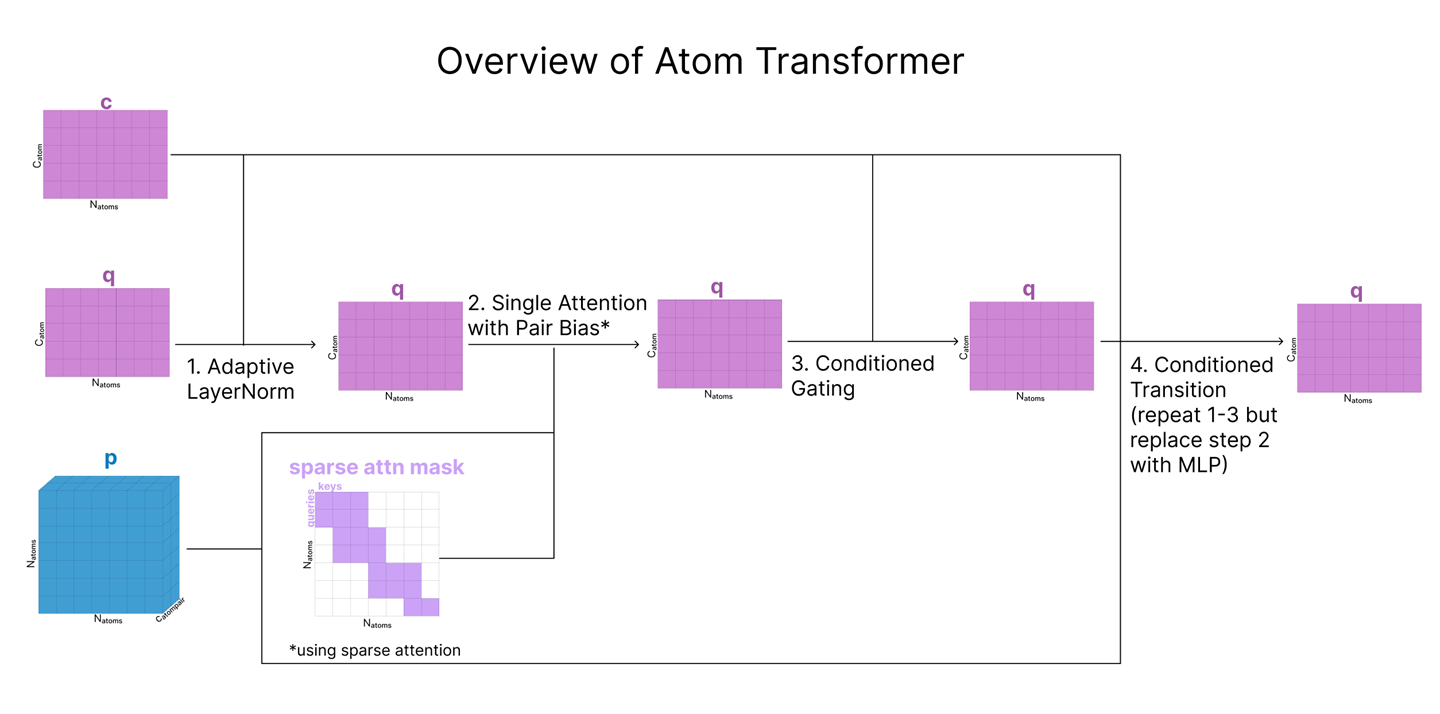
앞서 q(모든 원자를 표현하는 single)와 p(원자 쌍을 표현하는 pair)를 생성했습니다. 이제 주변 원자 정보를 활용해 이들을 업데이트해야 하는데, 이 때 Atom Transformer라는 모듈을 사용합니다. Atom Transformer는 여러 블록으로 구성되며, 각 블록에서는 p(pair 표현)와 q의 초기 사본( c )을 함께 참고하여, q를 갱신합니다. 이 때 c는 Attention Transformer에서 업데이트되지 않고 유지되므로, 시작 시점 표현을 “잔류(residual) 연결” 방식으로 계속 제공한다고 볼 수 있습니다.
Atom Transformer는 전반적으로 표준 Transformer 구조(레이어 정규화→어텐션→MLP)를 따르지만, c와 p(이차적 입력, “컨디셔닝” 역할)를 추가로 반영한다는 점이 특이합니다. 또한 Attention 후에 별도의 ‘게이트(gating)’ 단계를 삽입해, 모델이 배운 정보를 어느 정도 반영할지 선택하도록 합니다.
다음으로 이 4단계를 순서대로 살펴봅니다:
^ 즉, “초기 사본”이라 함은 “q가 업데이트되기 전에, 만들자마자 복제해 둔 q의 원본” 정도로 이해하면 됩니다.
1. Adaptive LayerNorm
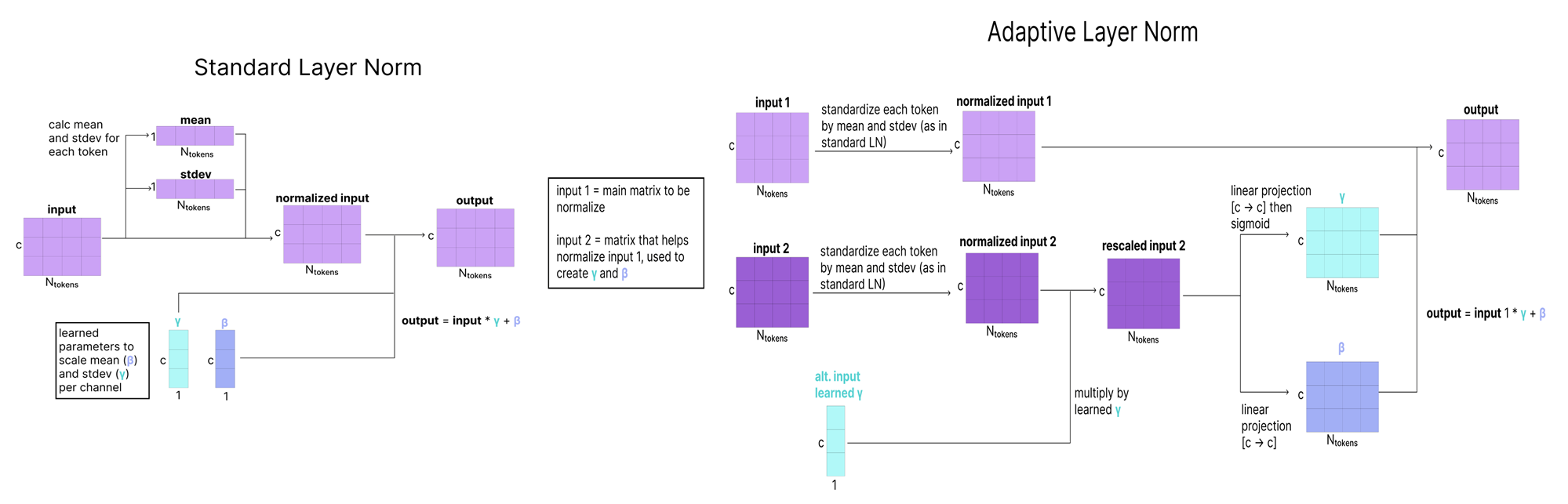
Adaptive LayerNorm (AdaNorm):
- AdaNorm은 이를 확장하여, 이 파라미터(γ, β)를 두 번째 입력(즉, Atom Transformer에서는 c)을 바탕으로 동적으로 예측합니다.
- 즉, 단순 고정값이 아니라, c에 따라 “q의 평균·표준편차를 어떻게 보정할지”가 결정되는 방식입니다.
- 기존 LayerNorm은 입력 행렬(예: q)에 대해, 채널별 스케일(γ)과 바이어스(β)를 학습된 상수로 적용합니다.
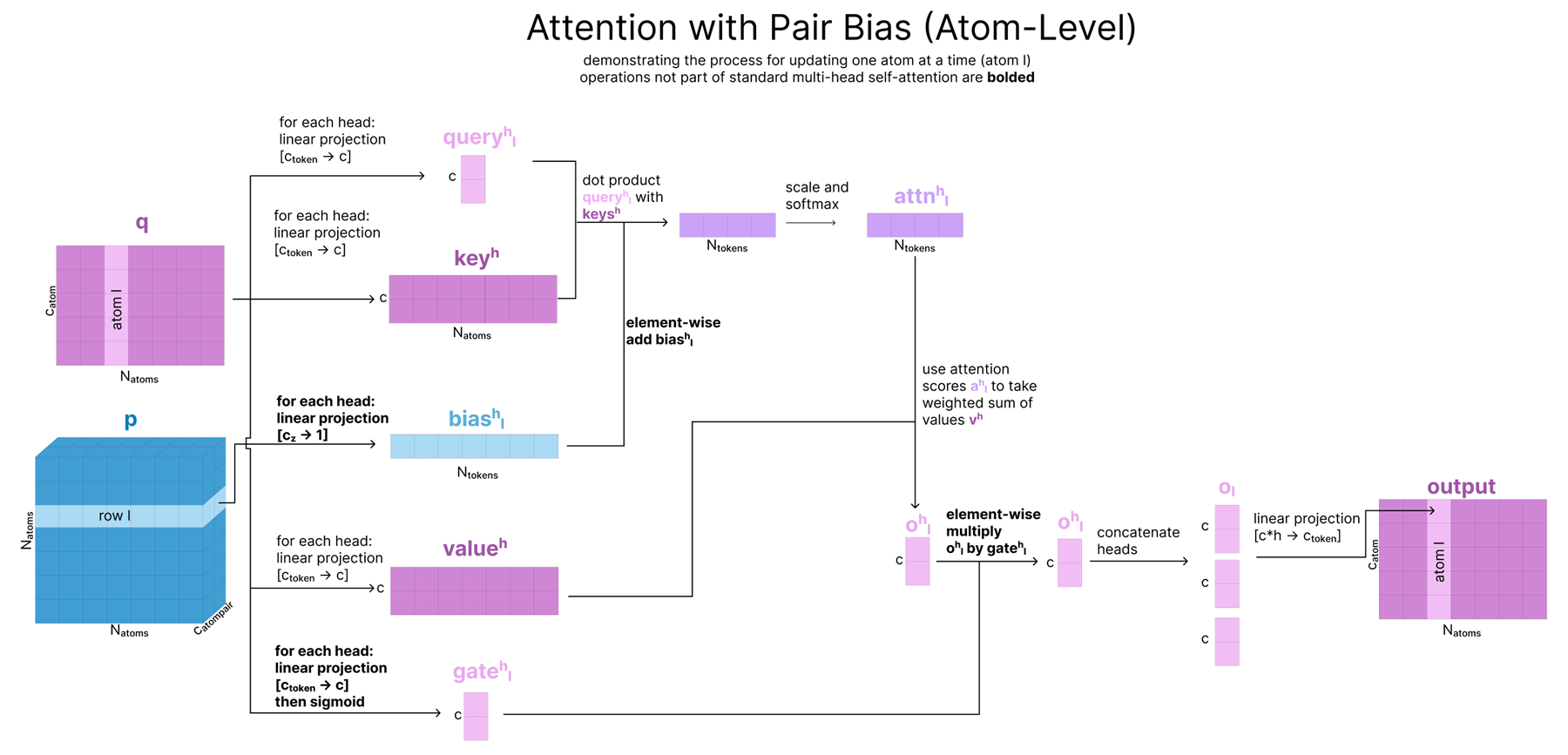
2. Attention with Pair Bias
원자 단위 어텐션(Atom-level Attention)은, 말 그대로 q(원자 시퀀스)에서 쿼리(query), 키(key), 밸류(value)를 뽑아 Self-Attention을 수행합니다.
다만, 여기에는 세 가지 중요한 차이가 있습니다:
- Pair-Biasing:
- 쿼리·키 내적(dot product) 결과에, pair 표현(p)에서 선형 변환한 값을 ‘바이어스’로 더해, 어텐션 가중치를 조정합니다.
- 이를 통해, 원자 쌍 간 결합 강도가 높은(p에서 강조) 원자들끼리는 어텐션이 더 크게 작동합니다.
- 단, 이 과정은 p가 q로부터 업데이트되는 것이 아니라, “p→q로 단방향으로” 영향을 주는 것임에 유의하세요.
- Gating:
- 쿼리·키·밸류 외에, q를 추가로 한 번 더 투영해 시그모이드(0~1 범위)로 통과시키는 “게이트”를 만듭니다.
- 어텐션 결과를 최종적으로 합칠 때, 이 게이트로 결과를 곱해주어 “어느 정도 정보만 반영”하도록 합니다.
- 이는 LSTM의 게이트와 비슷한 아이디어로, Residual 스트림에 실질적으로 어느 부분을 남길지 선택하게 해 줍니다.
- Sparse Attention:
- 원자 수가 많을 수 있으므로, 모든 원자→모든 원자 풀 어텐션을 돌리면 연산량이 매우 큽니다.
- 대신 “Sequence-local atom attention”이라는 스파스 어텐션을 써서, 32개 원자 단위 그룹이 한 번에 128개의 다른 원자를 주로 ‘참조(attend)’하는 식으로 제한합니다.
- 이는 최근 대용량 모델에서 흔히 사용하는 스파스 어텐션 기법과 유사합니다.
3. Conditioned Gating
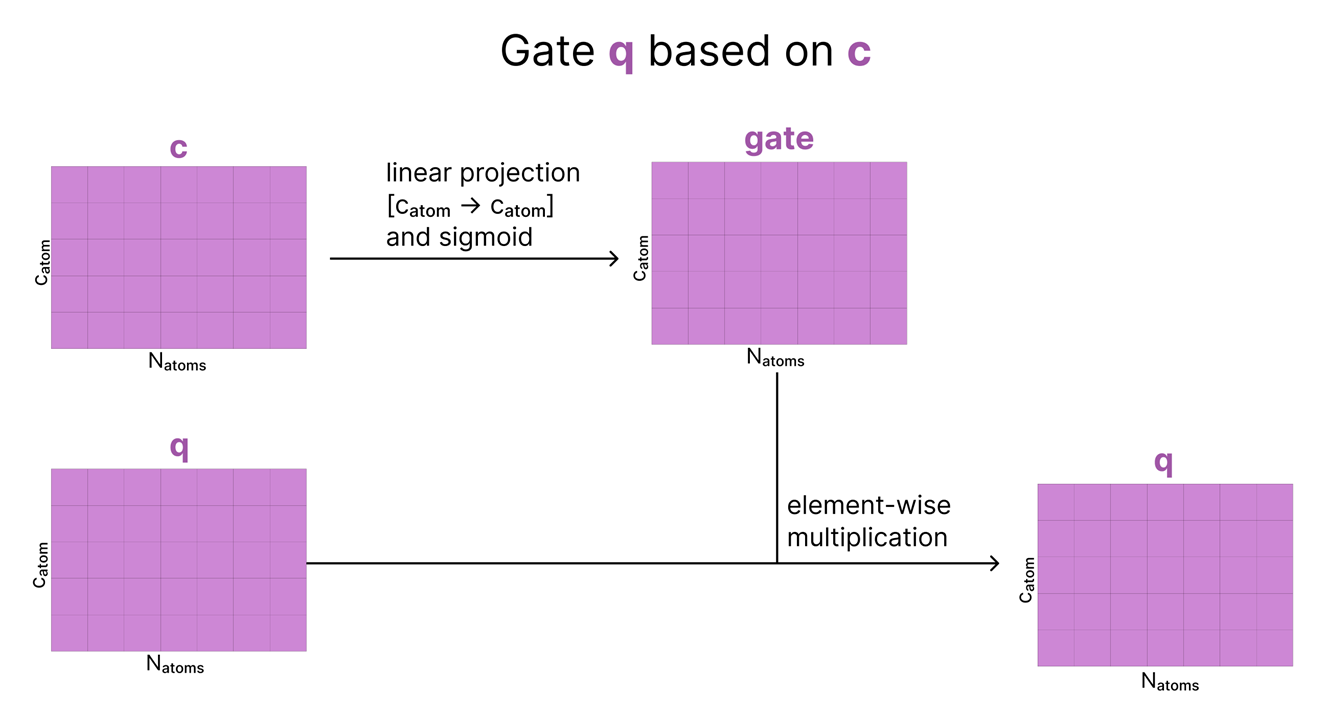
- 앞서 “Attention → 결과” 후에 한 번 게이트를 적용했지만, 이번에는 또 다른 게이트를 추가로 적용합니다.
- 다만 이번 게이트는 c(처음 원자 단위 single 표현)로부터 만들어져, 다시 한 번 “현재 결과에 얼마만큼 정보를 반영할지”를 결정합니다.
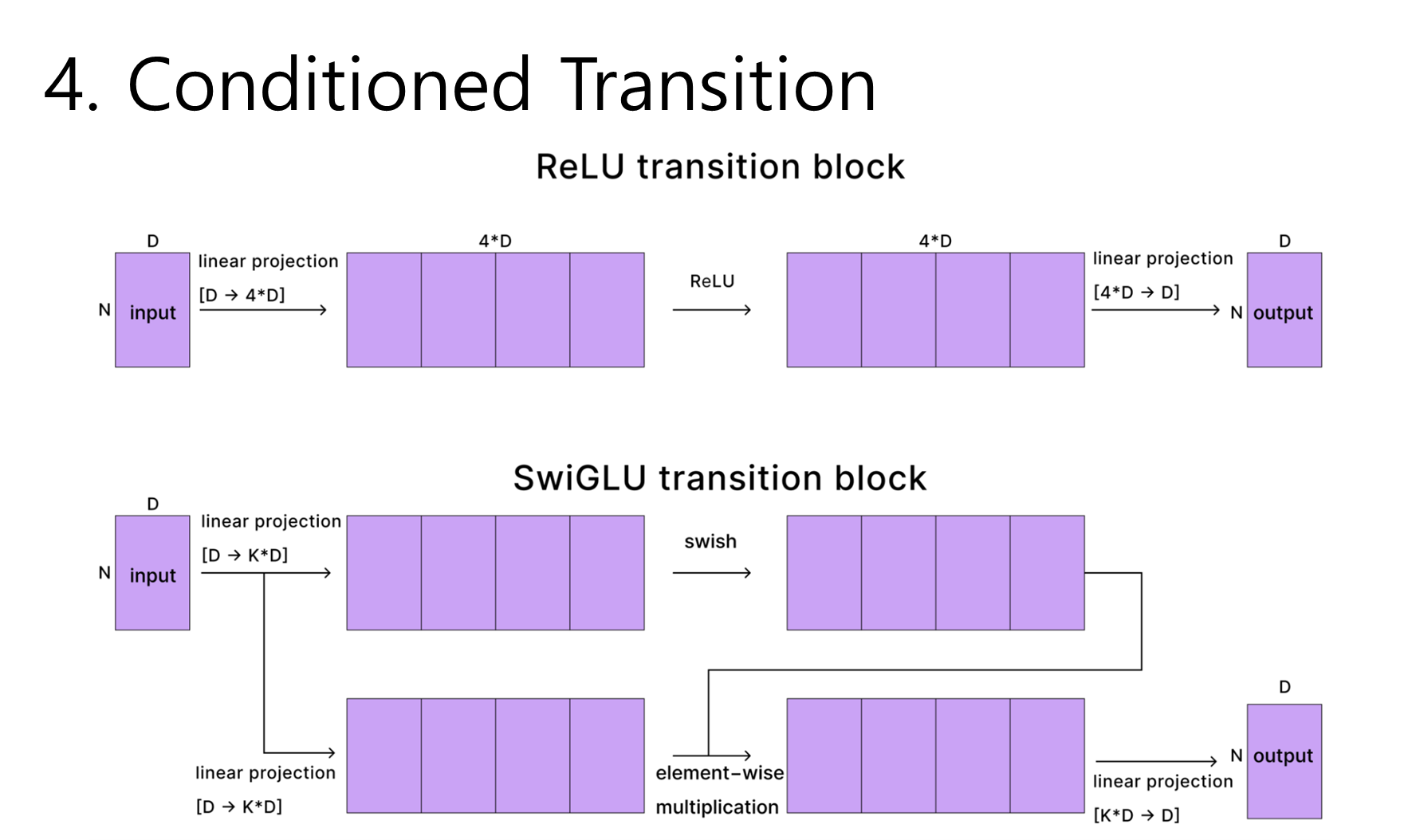
4. Conditioned Transition
- Transformer MLP 블록과 동일한 역할이지만, 여기서도 Adaptive LayerNorm(c 기반) + Conditional Gating(역시 c 기반)으로 감싸져 **“조건부(Conditioned)”**가 된다는 점이 특징입니다.
- SwiGLU 활성함수를 사용:
- AF2에서는 ReLU 기반 Transition을 썼지만, AF3에서는 SwiGLU로 바뀌었습니다.
- ReLU 대비 Swish(또는 SiLU) 함수가 곱해져 더 부드러운(non-linear) 반응을 제공하며, 최근 많은 신경망 아키텍처가 채택하는 방식입니다.
- 구체적으로 ReLU 방식은 (채널 4배 증폭)→(ReLU)→(원래 채널로 다운 프로젝트) 흐름이었으나, SwiGLU에서는 **(2개 분기 중 하나만 Swish 통과)→(결과를 곱)→(다운 프로젝트)**로 진행합니다.
정리하자면, Atom Transformer 블록은 위 4단계(Adaptive LayerNorm → Attention with Pair Bias → Conditioned Gating → Conditioned Transition)를 순서대로 수행하여, q(원자 단위 single)를 점진적으로 업데이트합니다. 이 과정에서 p(pair)와 c(초기 원자 표현)는 “조건부 입력(conditional input)”로 작동하여, 원자 간 상대관계나 초기 특성을 적절히 반영하도록 돕습니다.
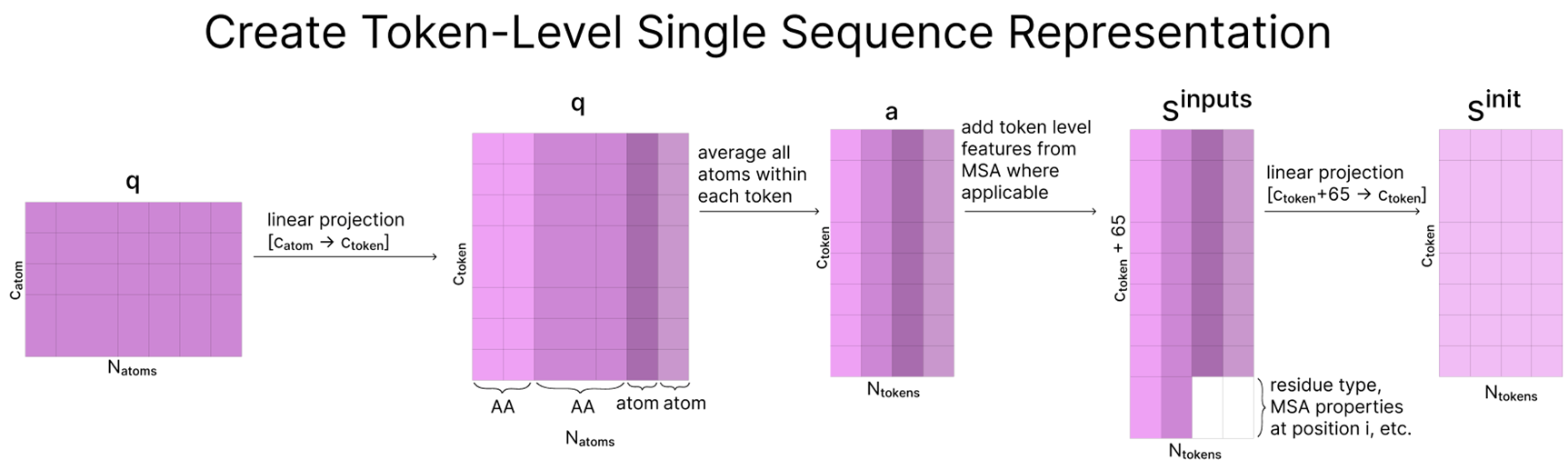
Aggregate Atom-Level → Token-Level
핵심 요약
- 원자 수준 → 토큰 수준으로 전환
- 지금까지 AF3는 모든 데이터를 “원자 단위”로 저장했지만, 표현 학습(representation learning) 단계부터는 “토큰 단위”로 작업합니다.
- “토큰”이란, 보통 아미노산 하나(표준 단백질)나 뉴클레오타이드 하나(표준 핵산)에 해당합니다.
- 토큰 단위 표현 만들기
- 원래 원자 단위 표현(128차원)을 더 큰 차원(384차원)으로 먼저 늘립니다.
- “같은 토큰에 속하는 원자들”의 표현을 평균하여, 토큰 한 개에 대응되는 벡터를 만듭니다.
- 이 규칙은 표준 아미노산/뉴클레오타이드에만 적용되며, 비표준 분자 등은 이미 “원자=토큰” 형태라 변화가 없습니다.
- MSA 정보와 결합 (sinputs)
- 토큰 단위 표현에 MSA(다중 서열 정렬)에서 얻은 특징·통계를 결합(concatenate) 합니다(있는 경우에만).
- 이렇게 합쳐진 행렬을 sinputs라 부릅니다.
- sinit 초기화
- sinputs가 너무 커졌으므로, 다시 384차원으로 투영해 sinit를 만듭니다.
- sinit: 모델이 다음 “표현 학습” 단계에서 실제로 업데이트할 토큰 단위 시퀀스 표현.
- 한편, sinputs는 나중에 “구조 예측(Structure Prediction)” 단계에서 쓰기 위해 따로 저장해 둡니다.
정리하자면, “원자 단위 정보”를 모아 “토큰 단위”로 바꾼 뒤, MSA 등 부가 정보를 합쳐서 sinit라는 초기 시퀀스 표현을 만든다는 과정입니다. 이후 모델의 본격적 학습(Representation Learning)에서 sinit가 계속 업데이트되고, 구조 예측 단계에서 sinputs가 다시 활용됩니다.
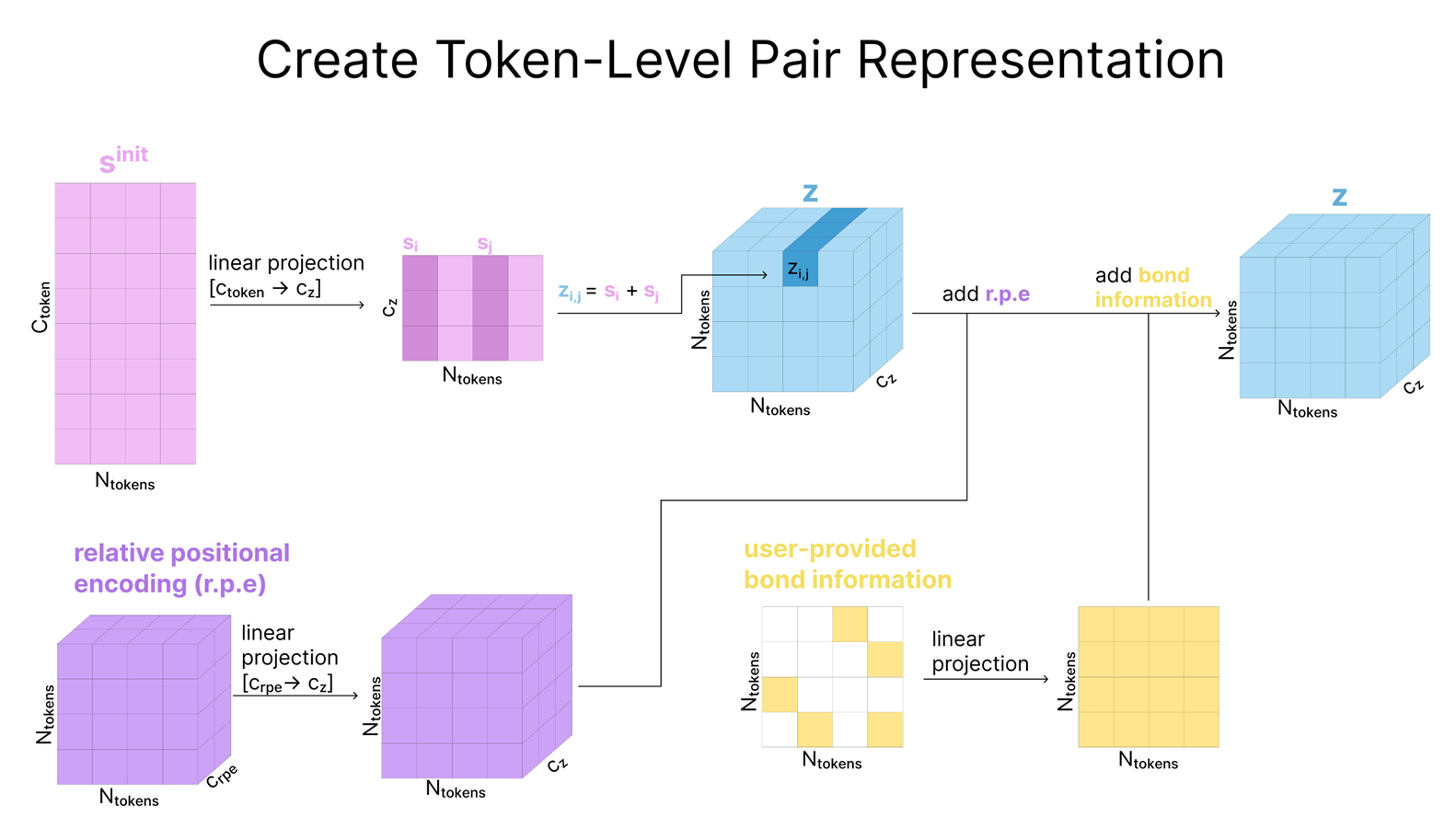
이 다이어그램은 토큰 단위 Single 표현(sinit)을 어떻게 Pair 표현(z)로 확장하고, 위치 인코딩과 사용자 지정 결합 정보 등을 어떻게 추가하는지를 단계별로 보여줍니다.
2단계에서는 원자 단위 표현(c, q, p)은 잠시 제쳐 두고, 다음 섹션에서 (m과 t의 도움을 받아) 토큰 단위 표현(s와 z)을 업데이트하는 데 집중할 것입니다.
이 부분은 내용이 너무 길고 어려운 내용이 많아서 가장 중요한 부분인 MSA Module에 대해서만 보겠습니다.
해당 섹션은 크게 Template module, MSA module, Pairformer, 반복(Repeated Blocks & Recycling) 이렇게 4가지로 나뉩니다.
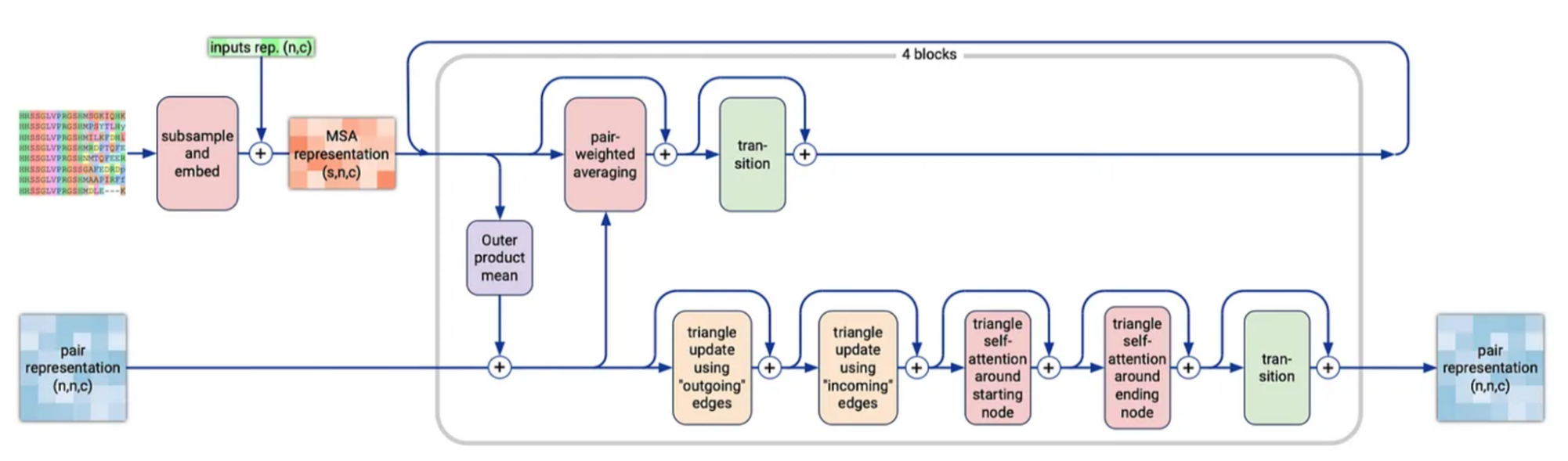
MSA module
이 MSA 모듈은 AlphaFold2의 Evoformer와 매우 유사하며, 핵심 목표는 MSA 표현과 pair 표현을 동시에 개선하면서, 양쪽이 상호작용(cross-talk)할 수 있도록 하는 것입니다.
구체적으로는, 먼저 MSA와 pair 텐서를 각각 독립적으로 업데이트하는 여러 연산을 수행한 뒤, 이 두 표현 간에 정보를 교환하도록 구성되어 있습니다.
가장 먼저 이루어지는 단계는, 이전에 생성된 MSA가 최대 16k 행(row)처럼 매우 클 수 있으므로, 일부 행만 샘플링(subsample)하여 사용한다는 점입니다. 그리고 이렇게 샘플링된 MSA 행들에, single 표현(s)을 선형 변환한 버전을 추가로 더해 줍니다(병합). 이를 통해 MSA와 단일 시퀀스 정보가 조금 더 밀접하게 연결된 상태에서 이후 업데이트가 이루어지도록 하는 것입니다.
그림에서 보면, “inputs rep. (n,c)” 라고 표시된 초록색 박스(혹은 화살표)가 바로 단일 시퀀스 표현(s)이 MSA와 결합될 때 들어오는 경로입니다. 다이어그램상에서는 이 선이 직접적으로 “subsample and embed” 블록(분홍색)에 더해져 MSA representation으로 이동하는 모습을 보여주는데,
- 이 초록색 입력이 곧 s(단일 표현)을 받아 MSA 채널과 맞추기 위한 선형 변환(Projection) 과정을 내포하고 있다고 보시면 됩니다.
- 다이어그램은 간단히 “inputs rep. (n,c) + subsample and embed”라고만 표기했지만, 실제 연산 내부에서는 s를 적절히 투영(채널 매핑)하여 MSA 행렬과 합(add)하는 작업이 수행됩니다.
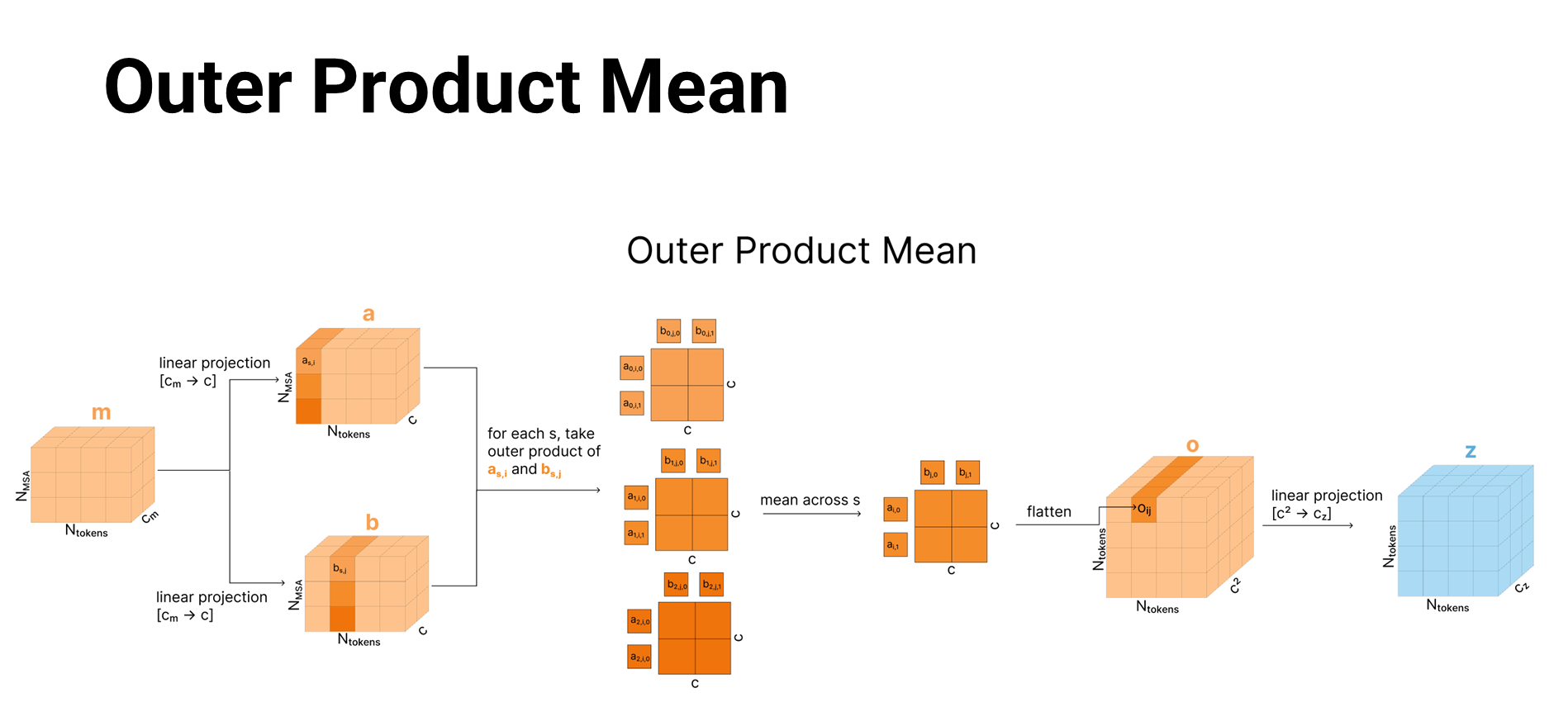
Outer Product Mean
Outer Product Mean 과정을 간단히 풀어 쓴 설명입니다:
- MSA에서 열(column) 2개를 비교
- MSA에서 두 위치(토큰 인덱스 i, j)를 잡으면, 각 열은 여러 개의 진화적 서열(행)을 가지고 있습니다.
- 이 두 열(ms,i와 ms,j)을 서로 비교하면, “서열 전체에서 두 위치가 어떻게 함께 변이했는지(공변·상관)” 같은 정보를 알 수 있게 됩니다.
- 각 진화적 서열마다 외적(outer product)을 구하고 평균
- 모든 진화적 서열을 순회하면서, (ms,i)와 (ms,j)에 대한 외적을 구합니다.
- 이어서 이 외적들을 평균(mean) 내어, i와 j 사이를 대표하는 하나의 결과를 얻게 됩니다.
- 외적은 “(채널 수)×(채널 수)” 형태인데, 이를 나중에 펼쳐(flatten)서 다시 원하는 차원으로 투영(Projection)합니다.
- Pair 표현 zi,j에 반영
- 이렇게 얻은 외적 평균 결과를 zi,에 더합니다.
- 즉, “두 위치(i, j) 간의 관계 정보”를 MSA에서 끌어와 pair 표현에 포함시키는 셈입니다.
- 의의
- 이 단계가 “진화적 서열들 간 교차 정보”를 서로 섞어주는 유일한 지점입니다.
- 덕분에 AF2의 Evoformer에서보다 계산량을 크게 줄이면서도, i와 j 사이의 상관관계를 충분히 활용할 수 있게 했습니다.
- AF2에 비해 MSA→Pair 정보 반영을 단순화하여 효율을 높인 것이 특징입니다.
정리하면, Outer Product Mean은 “MSA의 각 열끼리(토큰 인덱스 i, j) 외적을 구해 평균을 내고, 그 결과를 pair 표현에 반영”함으로써, 두 위치의 공진화(covariation) 정보를 zi,j에 주입하는 핵심 연산입니다.
- 내적은 “두 사람이 어느 정도 비슷한 취향을 갖는가”를 점수 하나로 말해 주는 것이고,
- 외적은 “A 사람의 취향 항목(예: 음식, 영화, 음악, 책 등) 각각과 B 사람의 항목 각각이 어디서 얼마나 어울리는지”를 전부 표로 만들어 놓은 것처럼, 모든 항목 쌍별 상호작용을 담아내는 것이라고 생각하면 됩니다.
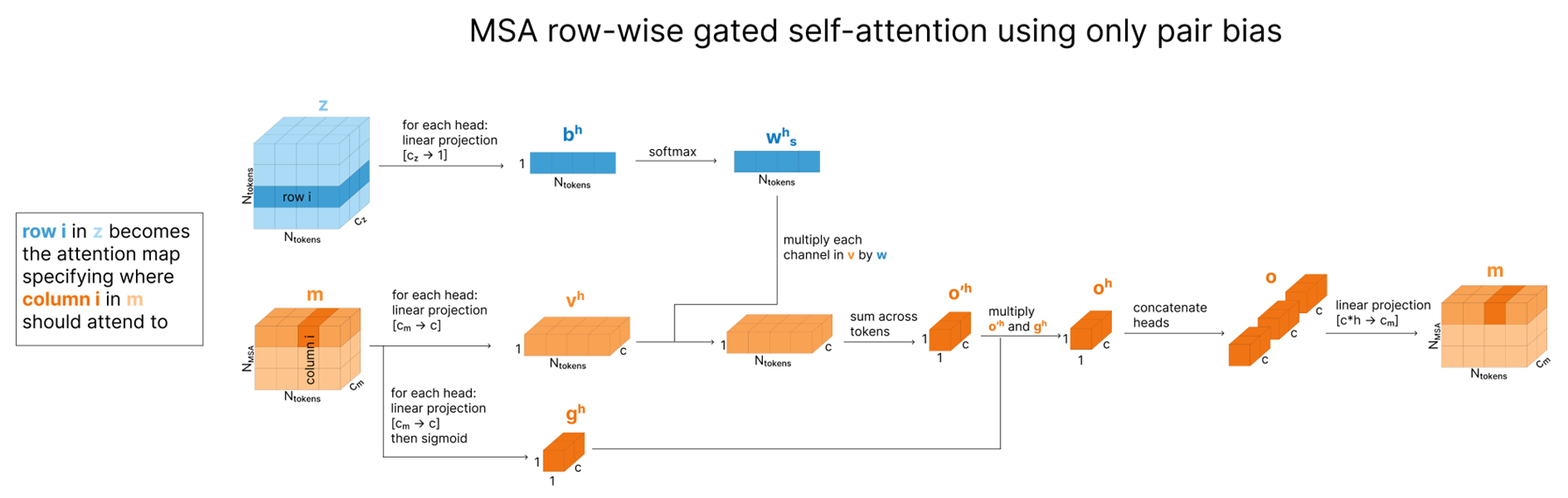
Row-wise gated self-attention using only pair bias
row-wise gated self attention using only pair bias는 MSA의 각 행(서열) 내에서 pair(z)가 제공하는 토큰 간 관계 정보를 바탕으로, 어떤 위치끼리 주목해야 하는지를 결정하고, 그 결과를 게이트로 조절하여 MSA 표현을 갱신합니다. 이는 MSA가 pair 표현에서 가져온 관계 정보를 자기 자신(MSA row) 안에 반영해, 최종적으로 MSA가 더욱 풍부한 맥락을 갖도록 하는 단계라고 볼 수 있습니다.
Structure Prediction
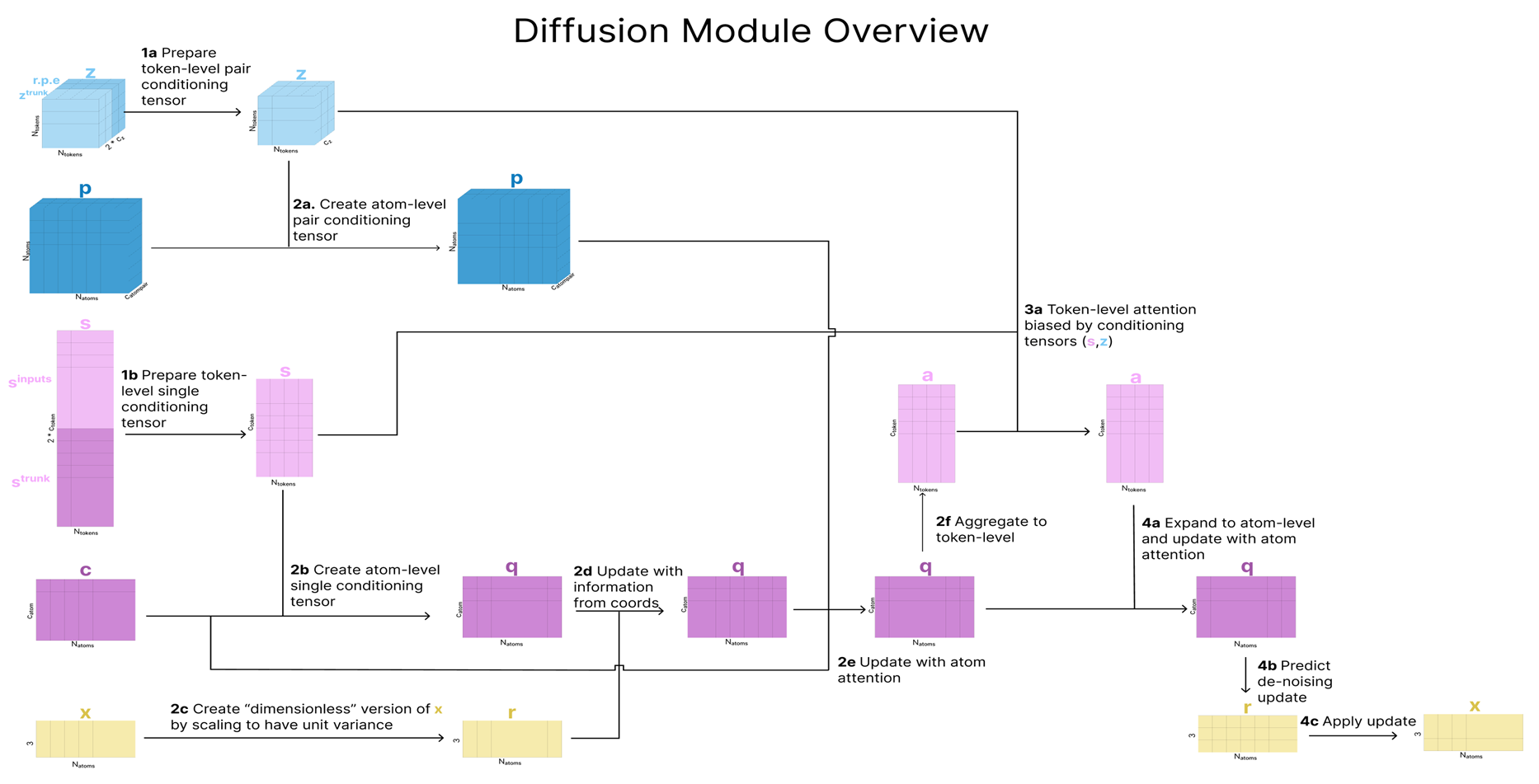
이제 구조 예측 섹션의 Diffusion 모델의 동작 방식을 보겠습니다.
이 섹션에서는, 토큰 수준에서 학습한 s, z 표현과 모델의 초기 원자·토큰 표현( c, p, sinputs 등)을 이용해, 실제 3D 좌표를 Diffusion 모델을 통해 생성합니다.
기본 원리
- Diffusion 모델은 데이터를 조금씩(여러 단계 t에 걸쳐) 무작위 노이즈로 덮어준 뒤, 모델이 “추가된 노이즈를 역으로 예측해서 제거”하도록 학습하는 방식입니다.
- 추론 시에는 완전히 노이즈로 뒤덮인 상태( xt=T )에서 시작해, 각 단계마다 모델이 예상하는 노이즈를 제거해가며, 최종적으로 깨끗한 상태(구조)를 얻습니다.
AF3에서의 Diffusion
- AF3가 학습하는 데이터( x )는 원자들의 (x, y, z) 좌표입니다.
- 학습 시에는 좌표에 가우시안 노이즈를 점차 많이 더하다가, 모델이 “추가된 노이즈”를 역추정하는 식으로 훈련합니다.
- 추론 시에는 무작위 좌표에서 시작해, 매 디퓨전 스텝마다 모델이 제시하는 노이즈를 제거해나가며 최종 구조를 얻습니다.
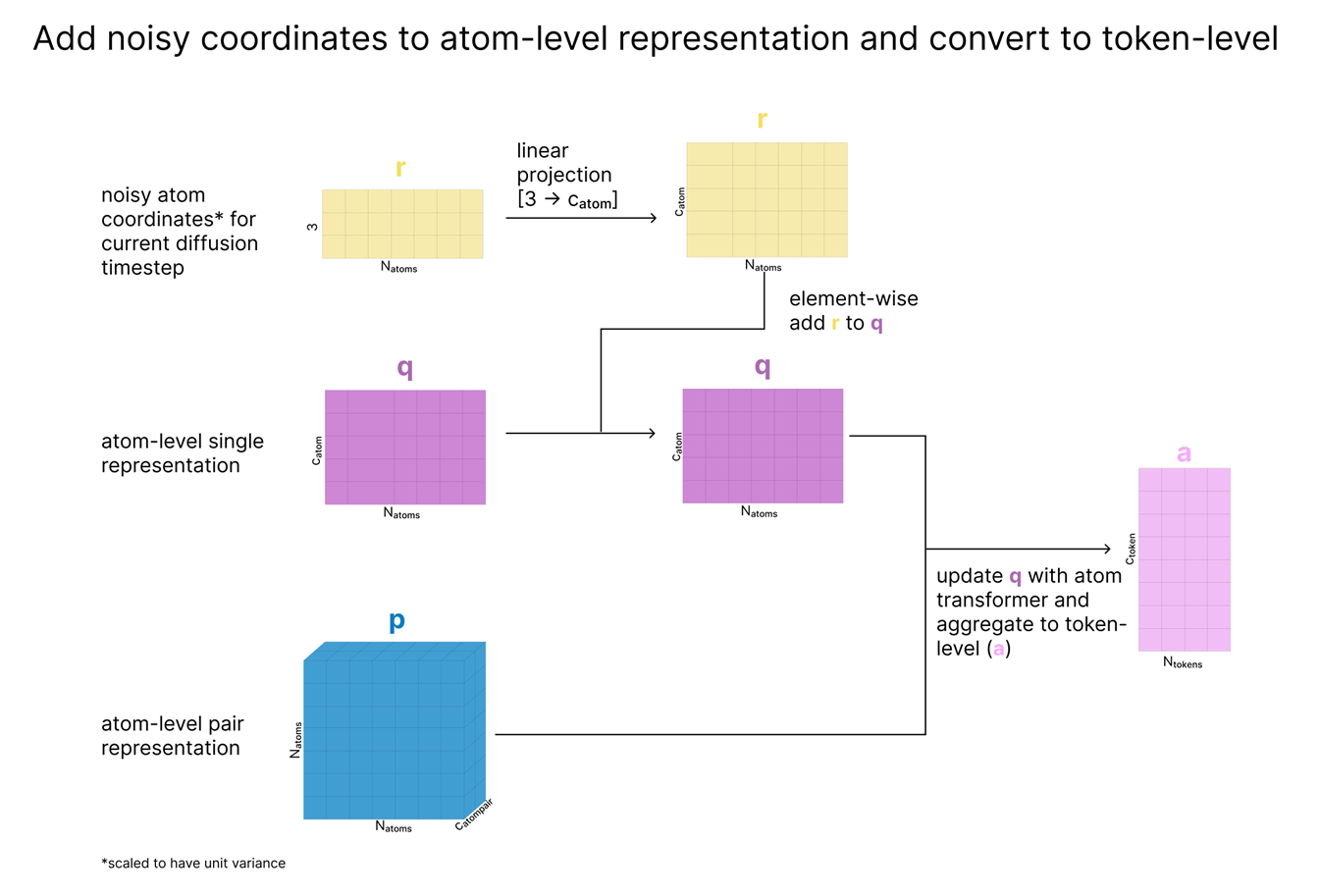
- AF3의 구조 예측은 완전히 “Atom-level 확산(diffusion)” 기법으로 새롭게 바뀌었습니다.
- 매 스텝마다 “원자 좌표를 노이즈화→모델이 노이즈를 예측해 제거” 과정을 거치며, 토큰 단위(s, z) 및 초기 원자 표현(c, p) 등의 정보를 조건(conditional)으로 활용합니다.
- 이 4단계(토큰→원자→토큰→원자) 루프를 거쳐, 최종적으로 안정적인 3D 구조를 출력하게 됩니다.
- 이전 AlphaFold2가 사용하던 “Invariant Point Attention” 대신, 디퓨전에서 “임의 회전·평행이동”을 매번 적용해 등가성을 유지하는 방식을 채택한 점도 중요한 변화입니다.
Insight
1. AlphaFold를 Retrieval-Augmented Generation(RAG)으로 해석하기
- AF2가 처음 나왔을 당시, 추론(inference) 중에 훈련 데이터(또는 이와 유사한)를 검색해 사용하는 방식은 흔하지 않았습니다.
- 예: MSA(다중 서열 정렬) & 템플릿 검색을 통해 구조 예측.
- 다른 딥러닝 분야(예: 이미지 분류)에서는 보통 추론 시 추가 검색을 하지 않는 경우가 많았습니다.
- AF3는 AF2에 비해 MSA 활용도를 줄이긴 했지만, 여전히 MSA와 템플릿을 추론 단계에서 사용합니다. (반면 ESMFold 등은 retrieval 없는 파라메트릭 접근)
- 최근에는 거대 언어모델(LLM)에서도 RAG 개념이 흔해졌습니다. 예: 검색 엔진 등을 활용해 최신 지식을 가져와 모델 추론에 반영.
결론: AF3가 MSA/템플릿을 사용해 예측하는 방식은, 대규모 언어모델에서의 RAG와 유사한 흐름이며, 향후 추론 시 “관련 예시 직접 참조” 접근이 더 확산될 가능성이 있습니다.
2. Pair-Bias Attention
- AF2, AF3에서 중요한 특징은 Pair-Bias가 들어간 어텐션입니다.
- 쿼리·키·밸류는 동일 소스로부터 오는 self-attention 형태이되, pair 표현에서 나온 추가 바이어스(term)를 주입해 “어떤 위치끼리 더 강하게 연결할지”를 결정.
- 이는 “가벼운 형태의 정보 공유”로 볼 수 있으며, 완전한 크로스 어텐션보다 부담이 적습니다.
- 단백질 분야에서는 Pair-Bias Attention이 다른 모델(예: RoseTTAFold 등)에도 일부 쓰이지만, 일반적인 NLP나 CV에서는 아직 흔치 않습니다.
3. Self-supervised 학습 이슈
- ESM 등은 “언어모델”처럼 자기지도(self-supervised) 방식으로 대규모 사전학습을 통해 MSA 없이도 구조 예측이 가능함을 보여주었습니다.
- AF2도 MSA 마스킹 예측을 일부 활용했으나, AF3에서는 그 부분이 제거되었습니다.
- 왜 자기지도 방식(예: MSA 기반 언어모델)으로 초기화하지 않았는지 작중에서 구체적으로 언급되지 않음.
- 가능성:
- 대규모 사전학습이 계산 낭비일 수 있다.
- 소규모 MSA 모듈을 유지하면, 사전학습 임베딩보다도 실시간 MSA가 더 성능이 낫다.
- AF3는 아미노산뿐 아니라 DNA/RNA/리간드(하이브리드 토큰)를 다루므로, 사전학습 임베딩과 결합하기 어려웠을 수 있다.
4. 분류(Classification) vs 회귀(Regression)
- AF2 시절부터, 거리 예측 시 일정 구간별 binning(분류)과 MSE 손실을 혼합 사용.
- 분류로 접근하면, 예측이 bin 하나만 벗어나도 오답 처리된다는 점에서 “근접 but off-by-one”을 충분히 보상 못 받을 수 있음.
- 그럼에도 불구하고, 분류 방식이 다양한 거리를 동시에 예측하는 데 있어 그레이디언트를 안정화한다는 실험적 이점이 있는 것으로 추정.
5. RNN 유사성(게이팅, 반복)과 재활용(Recycling)
- 게이트(gating)
- AF3 곳곳에 시그모이드 게이트를 두어, 정보가 Residual 스트림에 얼마나 반영될지 통제.
- 이는 LSTM이나 GRU 같은 재발생 신경망(RNN)의 게이팅과 유사하며, 일반적인 Transformer에는 흔치 않음.
- 반복 적용 및 가중치 재사용
- AF3는 동일 블록을 여러 번 반복(recycling)하며, 점진적으로 구조 예측을 개선.
- 이는 LSTM이 타임스텝마다 같은 가중치를 순환적으로 사용하는 것과 유사한 아이디어.
- 적응형 연산(adaptive computation)
- Recycling은 “입력이 어려우면 더 많은 스텝을 거치고, 쉬우면 덜 거치는” 식의 적응형 계산 체계와 맥이 닿음.
6. Cross-distillation
- AF3에서 low-confidence 영역 처리 시 AF2 예측 결과를 다시 활용(=교차 지식 증류).
- “이전 모델이 잘하던 부분을 새 모델이 못한다면, 그 부분만 이전 모델로부터 교정받자”라는 실용적 접근.
- 결과적으로 AF3는 RF Diffusion 같은 새로운 기술과 AF2의 장점을 결합하는 형태.
요약
AF3는 전반적으로 AF2의 성공 요소(Pair-bias Attention, Recycling)와 최근 ML 트렌드(RAG, 게이팅, 반복적 구조 개선)를 적극 반영하면서도, MSA의 비중을 줄이는 등 계산 효율과 확장성을 개선한 모델입니다. 이러한 발전 방향은 대형 언어모델의 RAG, LSTM-like 게이팅, 구조 예측에서의 분류 접근 등 여러 ML 아이디어와 맞닿아 있어, 향후 딥러닝 분야에서의 적용 범위가 더 확장될 가능성이 큽니다.